

Evolution and taxonomic classification of human papillomavirus 16 (HPV16)-related variant genomes: HPV31, HPV33, HPV35, HPV52, HPV58 and HPV67
- PMID: 21673791
- PMCID: PMC3103539
- DOI: 10.1371/journal.pone.0020183
Evolution and taxonomic classification of human papillomavirus 16 (HPV16)-related variant genomes: HPV31, HPV33, HPV35, HPV52, HPV58 and HPV67
Abstract
Background: Human papillomavirus 16 (HPV16) species group (alpha-9) of the Alphapapillomavirus genus contains HPV16, HPV31, HPV33, HPV35, HPV52, HPV58 and HPV67. These HPVs account for 75% of invasive cervical cancers worldwide. Viral variants of these HPVs differ in evolutionary history and pathogenicity. Moreover, a comprehensive nomenclature system for HPV variants is lacking, limiting comparisons between studies.
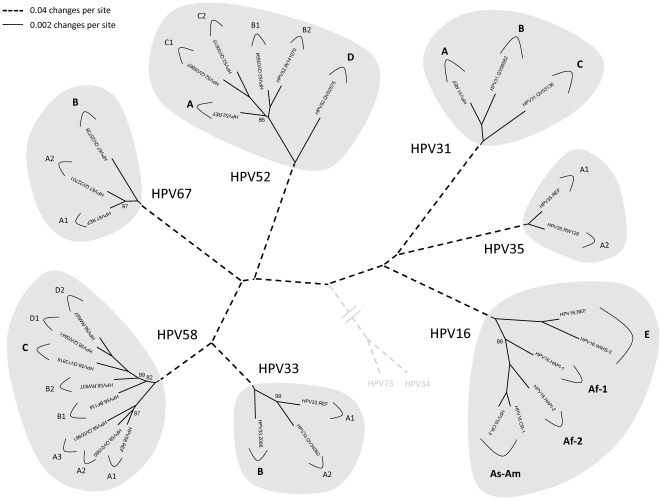
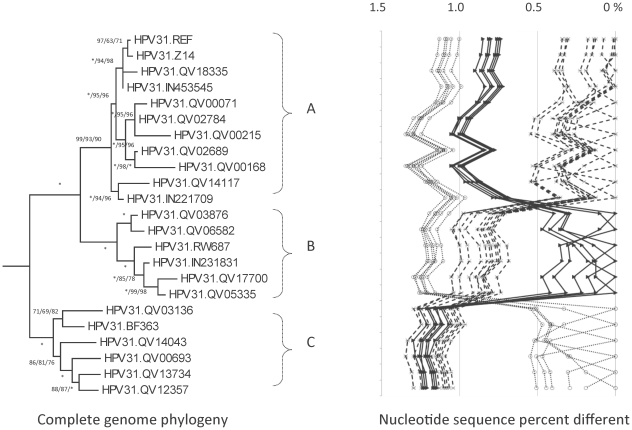
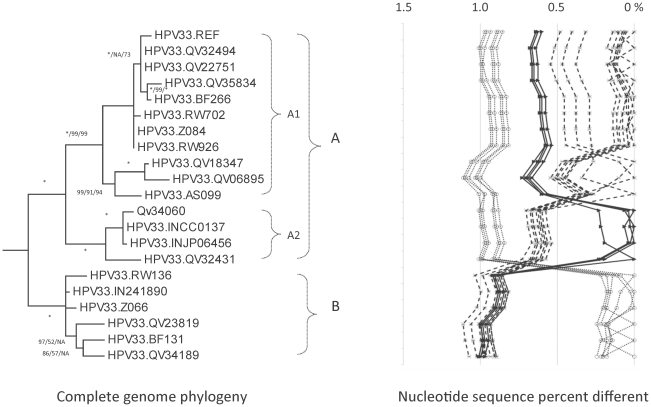
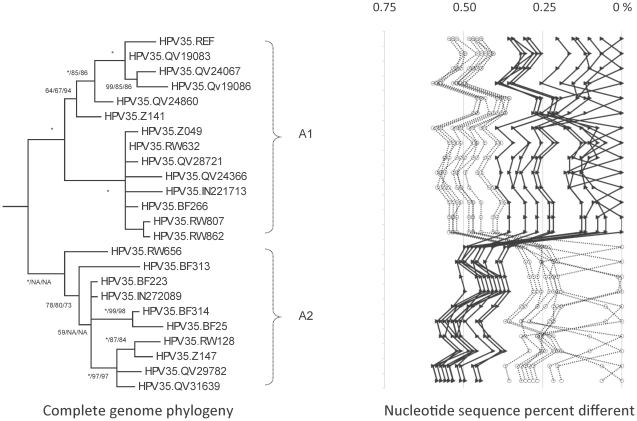
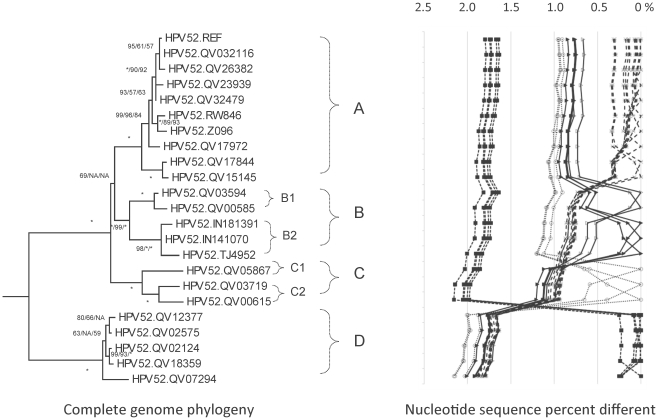
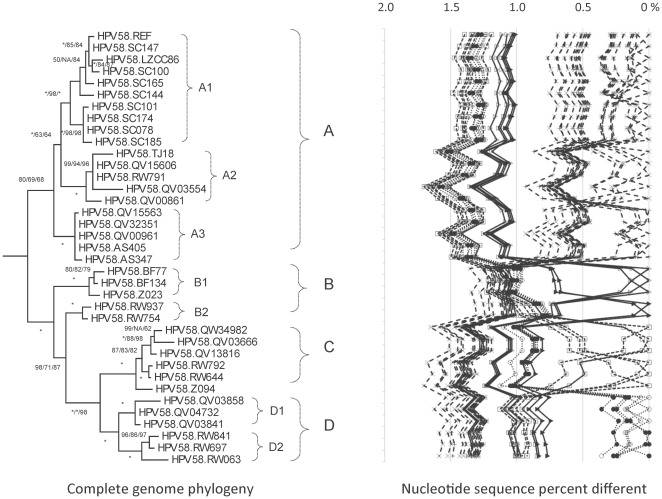
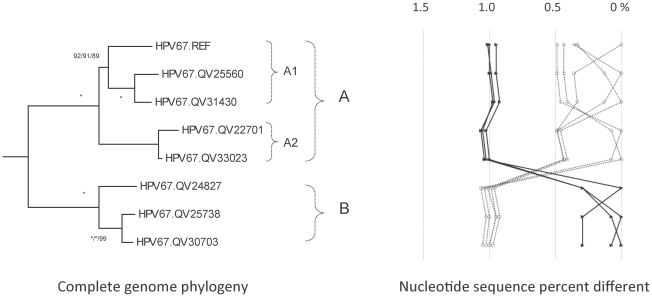
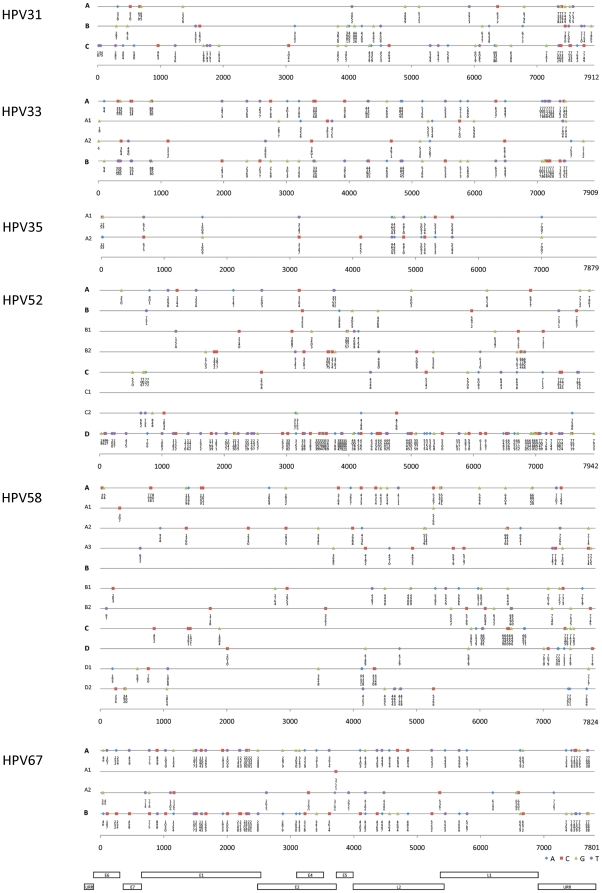
Methods: DNA from cervical samples previously characterized for HPV type were obtained from multiple geographic regions to screen for novel variants. The complete 8 kb genomes of 120 variants representing the major and minor lineages of the HPV16-related alpha-9 HPV types were sequenced to capture maximum viral heterogeneity. Viral evolution was characterized by constructing phylogenic trees based on complete genomes using multiple algorithms. Maximal and viral region specific divergence was calculated by global and pairwise alignments. Variant lineages were classified and named using an alphanumeric system; the prototype genome was assigned to the A lineage for all types.
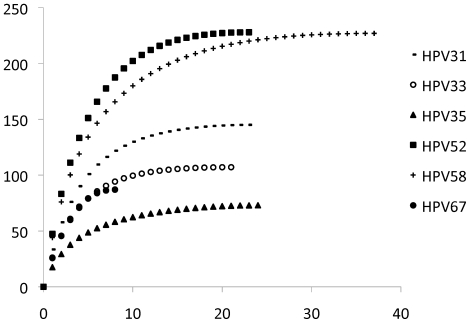
Results: The range of genome-genome sequence heterogeneity varied from 0.6% for HPV35 to 2.2% for HPV52 and included 1.4% for HPV31, 1.1% for HPV33, 1.7% for HPV58 and 1.1% for HPV67. Nucleotide differences of approximately 1.0% - 10.0% and 0.5%-1.0% of the complete genomes were used to define variant lineages and sublineages, respectively. Each gene/region differs in sequence diversity, from most variable to least variable: noncoding region 1 (NCR1) /noncoding region 2 (NCR2) >upstream regulatory region (URR)> E6/E7 > E2/L2 > E1/L1.
Conclusions: These data define maximum viral genomic heterogeneity of HPV16-related alpha-9 HPV variants. The proposed nomenclature system facilitates the comparison of variants across epidemiological studies. Sequence diversity and phylogenies of this clinically important group of HPVs provides the basis for further studies of discrete viral evolution, epidemiology, pathogenesis and preventative/therapeutic interventions.
Conflict of interest statement
Figures
References
-
- Jemal A, Bray F Center MM, Ferlay J, Ward E, et al. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. - PubMed
-
- Ferlay J, Shin HR, Bray F, Forman D, Mathers C, et al. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2011;127:2893–2917. - PubMed
-
- de Villiers EM, Fauquet C, Broker TR, Bernard HU, zur Hausen H. Classification of papillomaviruses. Virology. 2004;324:17–27. - PubMed
-
- Munoz N, Bosch FX, de Sanjose S, Herrero R, Castellsague X, et al. Epidemiologic classification of human papillomavirus types associated with cervical cancer. N Engl J Med. 2003;348:518–527. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
