

Whole-genome sequencing for optimized patient management
- PMID: 21677200
- PMCID: PMC3314311
- DOI: 10.1126/scitranslmed.3002243
Whole-genome sequencing for optimized patient management
Abstract
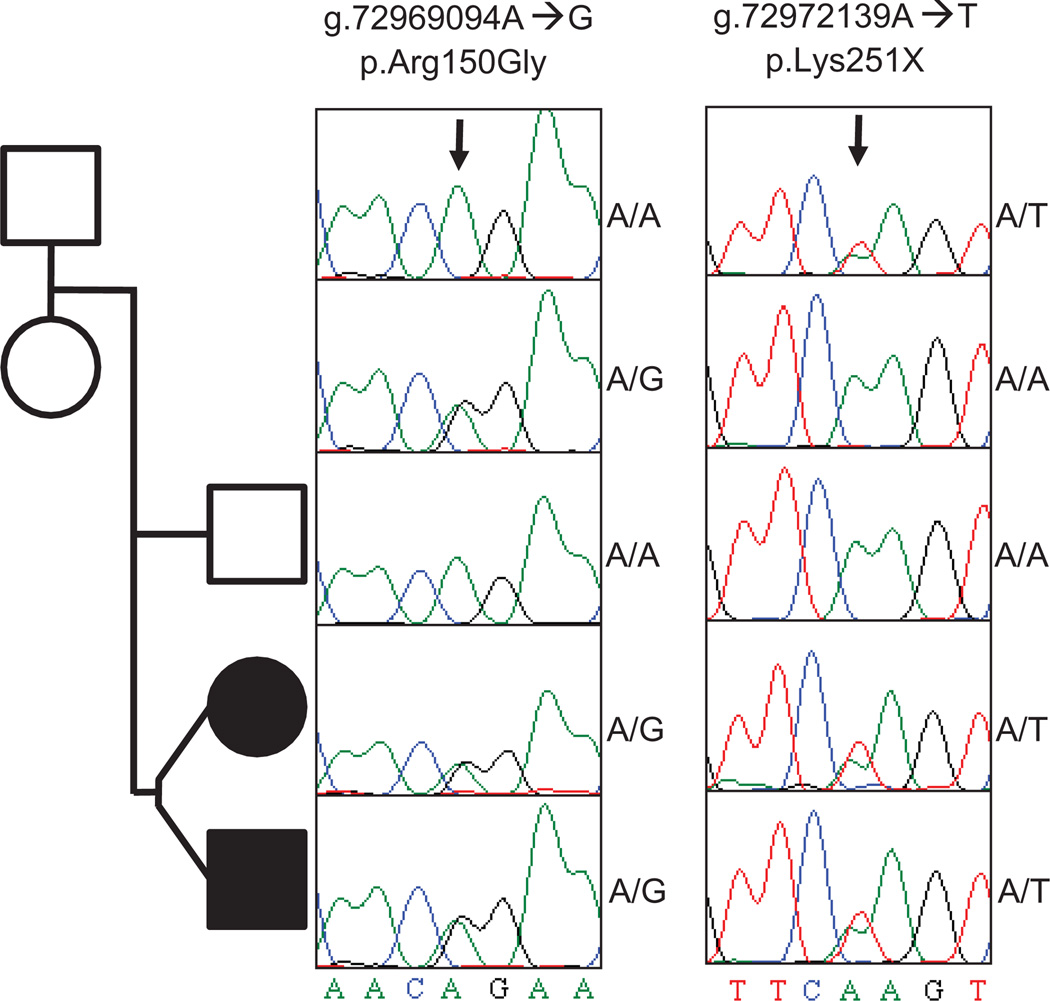
Whole-genome sequencing of patient DNA can facilitate diagnosis of a disease, but its potential for guiding treatment has been under-realized. We interrogated the complete genome sequences of a 14-year-old fraternal twin pair diagnosed with dopa (3,4-dihydroxyphenylalanine)-responsive dystonia (DRD; Mendelian Inheritance in Man #128230). DRD is a genetically heterogeneous and clinically complex movement disorder that is usually treated with l-dopa, a precursor of the neurotransmitter dopamine. Whole-genome sequencing identified compound heterozygous mutations in the SPR gene encoding sepiapterin reductase. Disruption of SPR causes a decrease in tetrahydrobiopterin, a cofactor required for the hydroxylase enzymes that synthesize the neurotransmitters dopamine and serotonin. Supplementation of l-dopa therapy with 5-hydroxytryptophan, a serotonin precursor, resulted in clinical improvements in both twins.
Figures


References
-
- Richards CS, Bale S, Bellissimo DB, Das S, Grody WW, Hegde MR, Lyon E, Ward BE Molecular Subcommittee of the ACMG Laboratory Quality Assurance Committee. ACMG recommendations for standards for interpretation and reporting of sequence variations: Revisions 2007. Genet. Med. 2008;10:294–300. - PubMed
-
- Fink JK, Ravin PD, Filling-Katz M, Argoff CE, Hallett M. Clinical and genetic analysis of progressive dystonia with diurnal variation. Arch. Neurol. 1991;48:908–911. - PubMed
-
- Nygaard TG, Marsden CD, Fahn S. Dopa-responsive dystonia: Long-term treatment response and prognosis. Neurology. 1991;41:174–181. - PubMed
-
- Geyer HL, Bressman SB. The diagnosis of dystonia. Lancet Neurol. 2006;5:780–790. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
