

Incidence, phenotypic features and molecular genetics of Kallmann syndrome in Finland
- PMID: 21682876
- PMCID: PMC3143089
- DOI: 10.1186/1750-1172-6-41
Incidence, phenotypic features and molecular genetics of Kallmann syndrome in Finland
Abstract
Background: Kallmann syndrome (KS), comprised of congenital hypogonadotropic hypogonadism (HH) and anosmia, is a clinically and genetically heterogeneous disorder. Its exact incidence is currently unknown, and a mutation in one of the identified KS genes has only been found in ~30% of the patients.
Methods: Herein, we investigated epidemiological, clinical, and genetic features of KS in Finland.
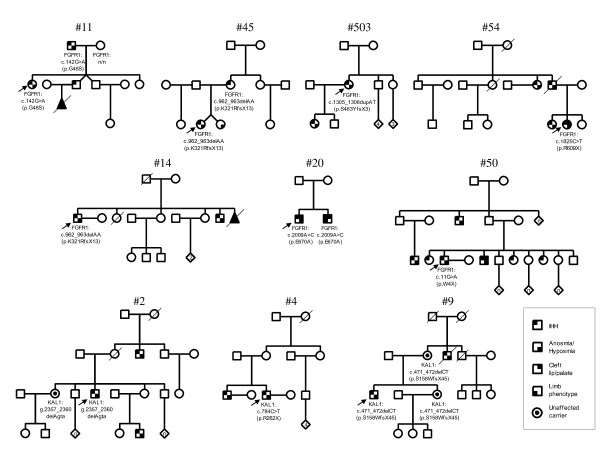
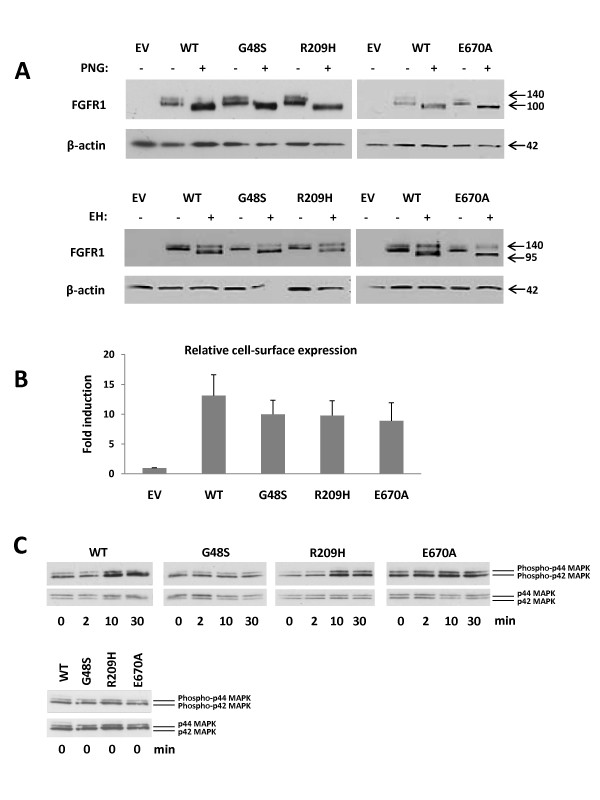
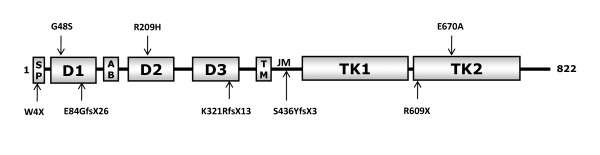
Results: The minimal incidence estimate of KS in Finland was 1:48 000, with clear difference between males (1:30 000) and females (1:125 000) (p = 0.02). The reproductive phenotype of 30 probands (25 men; 5 women) ranged from severe HH to partial puberty. Comprehensive mutation analysis of all 7 known KS genes (KAL1, FGFR1, FGF8, PROK2, PROKR2, CHD7, and WDR11) in these 30 well-phenotyped probands revealed mutations in KAL1 (3 men) and FGFR1 (all 5 women vs. 4/25 men), but not in other genes.
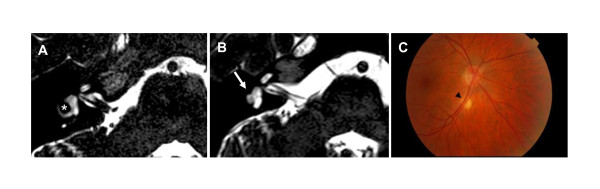
Conclusions: Our results suggest that Finnish KS men harbor mutations in gene(s) yet-to-be discovered with sex-dependent penetrance of the disease phenotype. In addition, some KS patients without CHD7 mutations display CHARGE-syndrome associated phenotypic features (e.g. ear or eye anomalies), possibly implying that, in addition to CHD7, there may be other genes associated with phenotypes ranging from KS to CHARGE.
Figures
References
-
- Schwanzel-Fukuda M, Bick D, Pfaff DW. Luteinizing hormone-releasing hormone (LHRH)-expressing cells do not migrate normally in an inherited hypogonadal (Kallmann) syndrome. Brain Res Mol Brain Res. 1989;6:311–326. - PubMed
-
- Raivio T, Sidis Y, Plummer L, Chen H, Ma J, Mukherjee A, Jacobson-Dickman E, Quinton R, Van Vliet G, Lavoie H, Hughes VA, Dwyer A, Hayes FJ, Xu S, Sparks S, Kaiser UB, Mohammadi M, Pitteloud N. Impaired fibroblast growth factor receptor 1 signaling as a cause of normosmic idiopathic hypogonadotropic hypogonadism. J Clin Endocrinol Metab. 2009;94:4380–4390. doi: 10.1210/jc.2009-0179. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
