

Expansion of intronic GGCCTG hexanucleotide repeat in NOP56 causes SCA36, a type of spinocerebellar ataxia accompanied by motor neuron involvement
- PMID: 21683323
- PMCID: PMC3135815
- DOI: 10.1016/j.ajhg.2011.05.015
Expansion of intronic GGCCTG hexanucleotide repeat in NOP56 causes SCA36, a type of spinocerebellar ataxia accompanied by motor neuron involvement
Abstract
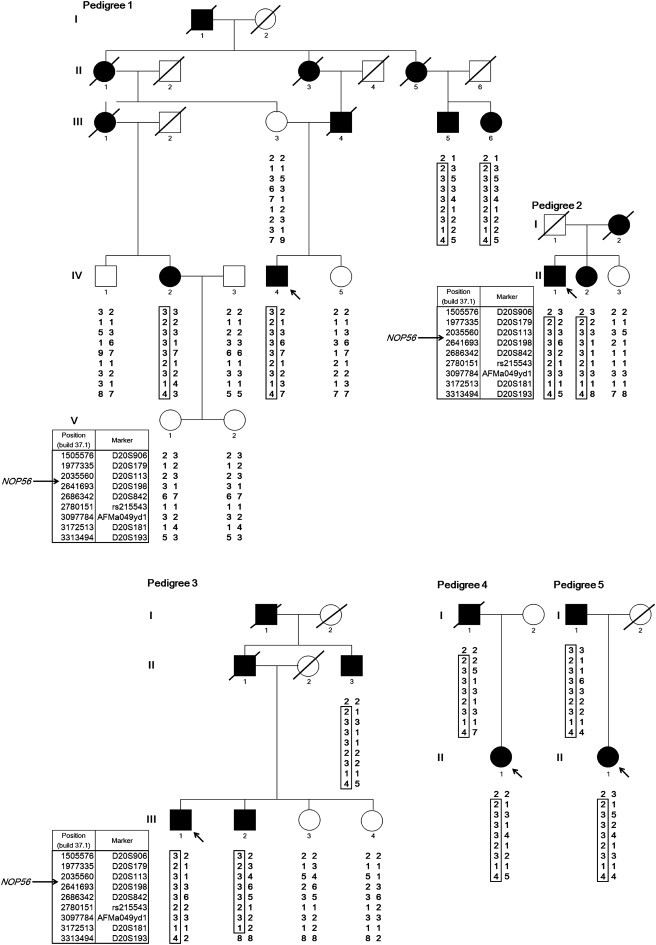
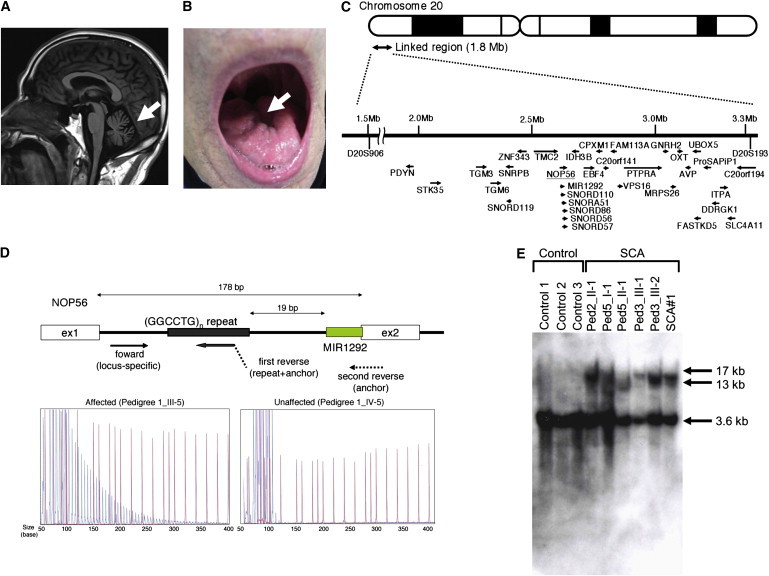
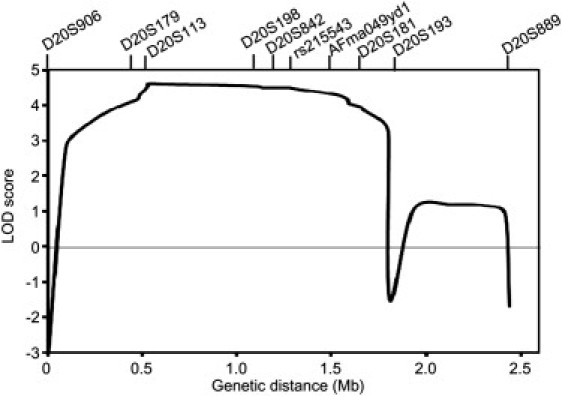
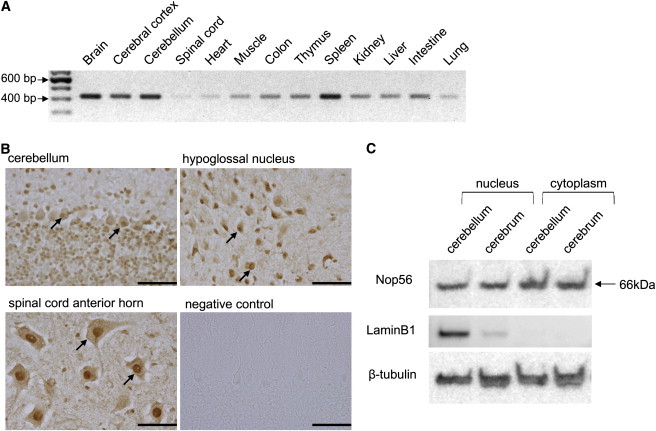
Autosomal-dominant spinocerebellar ataxias (SCAs) are a heterogeneous group of neurodegenerative disorders. In this study, we performed genetic analysis of a unique form of SCA (SCA36) that is accompanied by motor neuron involvement. Genome-wide linkage analysis and subsequent fine mapping for three unrelated Japanese families in a cohort of SCA cases, in whom molecular diagnosis had never been performed, mapped the disease locus to the region of a 1.8 Mb stretch (LOD score of 4.60) on 20p13 (D20S906-D20S193) harboring 37 genes with definitive open reading frames. We sequenced 33 of these and observed a large expansion of an intronic GGCCTG hexanucleotide repeat in NOP56 and an unregistered missense variant (Phe265Leu) in C20orf194, but we found no mutations in PDYN and TGM6. The expansion showed complete segregation with the SCA phenotype in family studies, whereas Phe265Leu in C20orf194 did not. Screening of the expansions in the SCA cohort cases revealed four additional occurrences, but none were revealed in the cohort of 27 Alzheimer disease cases, 154 amyotrophic lateral sclerosis cases, or 300 controls. In total, nine unrelated cases were found in 251 cohort SCA patients (3.6%). A founder haplotype was confirmed in these cases. RNA foci formation was detected in lymphoblastoid cells from affected subjects by fluorescence in situ hybridization. Double staining and gel-shift assay showed that (GGCCUG)n binds the RNA-binding protein SRSF2 but that (CUG)(6) does not. In addition, transcription of MIR1292, a neighboring miRNA, was significantly decreased in lymphoblastoid cells of SCA patients. Our finding suggests that SCA36 is caused by hexanucleotide repeat expansions through RNA gain of function.
Copyright © 2011 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Harding A.E. The clinical features and classification of the late onset autosomal dominant cerebellar ataxias. A study of 11 families, including descendants of the ‘the Drew family of Walworth’. Brain. 1982;105:1–28. - PubMed
-
- Matilla-Dueñas A., Sánchez I., Corral-Juan M., Dávalos A., Alvarez R., Latorre P. Cellular and molecular pathways triggering neurodegeneration in the spinocerebellar ataxias. Cerebellum. 2010;9:148–166. - PubMed
-
- Schöls L., Bauer P., Schmidt T., Schulte T., Riess O. Autosomal dominant cerebellar ataxias: clinical features, genetics, and pathogenesis. Lancet Neurol. 2004;3:291–304. - PubMed
-
- Ohta Y., Hayashi T., Nagai M., Okamoto M., Nagotani S., Nagano I., Ohmori N., Takehisa Y., Murakami T., Shoji M. Two cases of spinocerebellar ataxia accompanied by involvement of the skeletal motor neuron system and bulbar palsy. Intern. Med. 2007;46:751–755. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
