

PhyloCSF: a comparative genomics method to distinguish protein coding and non-coding regions
- PMID: 21685081
- PMCID: PMC3117341
- DOI: 10.1093/bioinformatics/btr209
PhyloCSF: a comparative genomics method to distinguish protein coding and non-coding regions
Abstract
Motivation: As high-throughput transcriptome sequencing provides evidence for novel transcripts in many species, there is a renewed need for accurate methods to classify small genomic regions as protein coding or non-coding. We present PhyloCSF, a novel comparative genomics method that analyzes a multispecies nucleotide sequence alignment to determine whether it is likely to represent a conserved protein-coding region, based on a formal statistical comparison of phylogenetic codon models.
Results: We show that PhyloCSF's classification performance in 12-species Drosophila genome alignments exceeds all other methods we compared in a previous study. We anticipate that this method will be widely applicable as the transcriptomes of many additional species, tissues and subcellular compartments are sequenced, particularly in the context of ENCODE and modENCODE, and as interest grows in long non-coding RNAs, often initially recognized by their lack of protein coding potential rather than conserved RNA secondary structures.
Availability and implementation: The Objective Caml source code and executables for GNU/Linux and Mac OS X are freely available at http://compbio.mit.edu/PhyloCSF CONTACT: mlin@mit.edu; manoli@mit.edu.
Figures
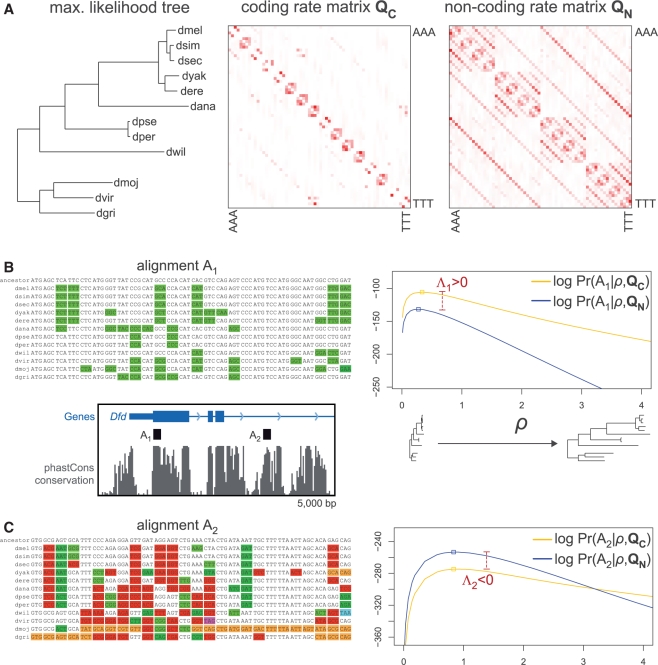
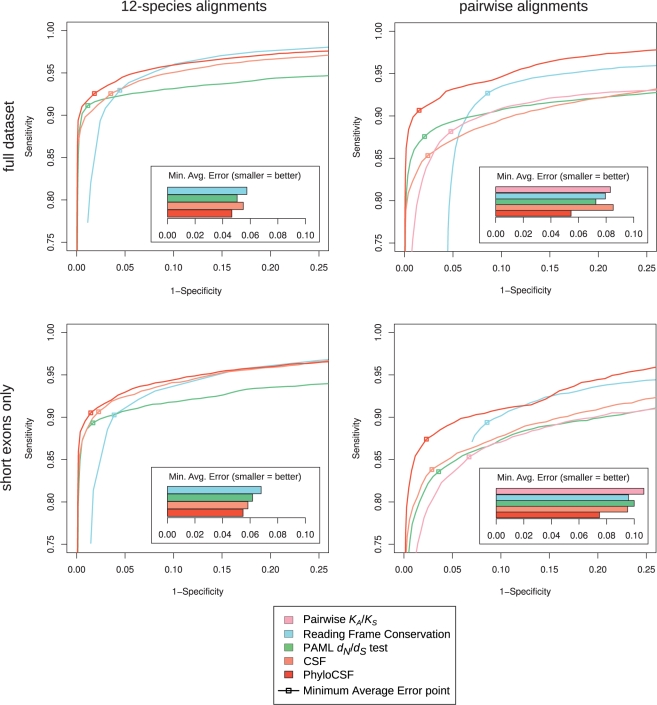
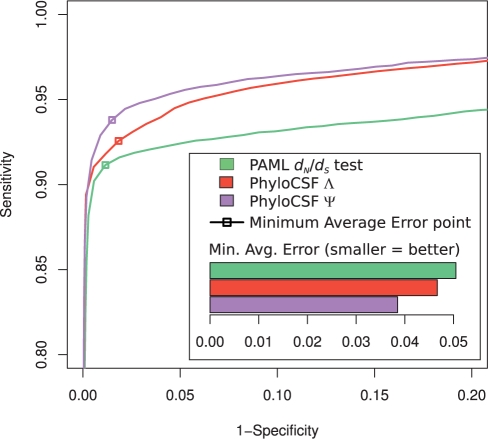
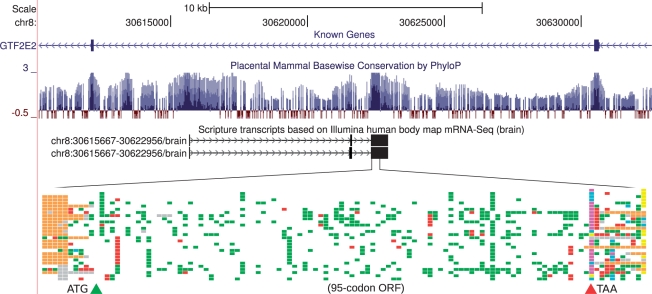
References
-
- Alioto T., Guigó R. State of the art in eukaryotic gene prediction. In: Frishman D., Valencia A., editors. Modern Genome Annotation: the BioSapiens Network. New York: Springer; 2009. pp. 7–40.
-
- Anisimova M., Kosiol C. Investigating protein-coding sequence evolution with probabilistic codon substitution models. Mol. Biol. Evol. 2008;26:255–271. - PubMed
-
- Arvestad L., Bruno W.J. Estimation of reversible substitution matrices from multiple pairs of sequences. J. Mol. Evol. 1997;45:696–703. - PubMed
-
- Brent M.R. Steady progress and recent breakthroughs in the accuracy of automated genome annotation. Nat. Rev. Genet. 2008;9:62–73. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
