

A novel computational framework for simultaneous integration of multiple types of genomic data to identify microRNA-gene regulatory modules
- PMID: 21685098
- PMCID: PMC3117336
- DOI: 10.1093/bioinformatics/btr206
A novel computational framework for simultaneous integration of multiple types of genomic data to identify microRNA-gene regulatory modules
Abstract
Motivation: It is well known that microRNAs (miRNAs) and genes work cooperatively to form the key part of gene regulatory networks. However, the specific functional roles of most miRNAs and their combinatorial effects in cellular processes are still unclear. The availability of multiple types of functional genomic data provides unprecedented opportunities to study the miRNA-gene regulation. A major challenge is how to integrate the diverse genomic data to identify the regulatory modules of miRNAs and genes.
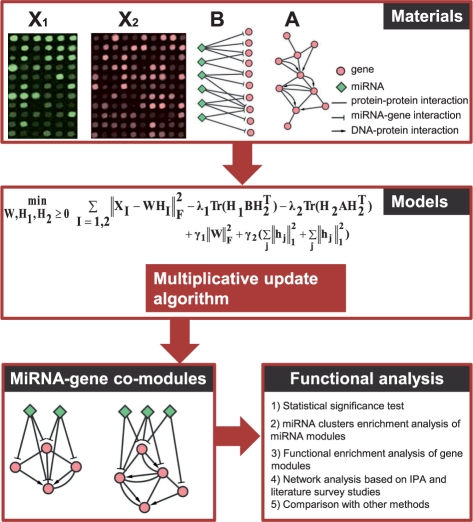
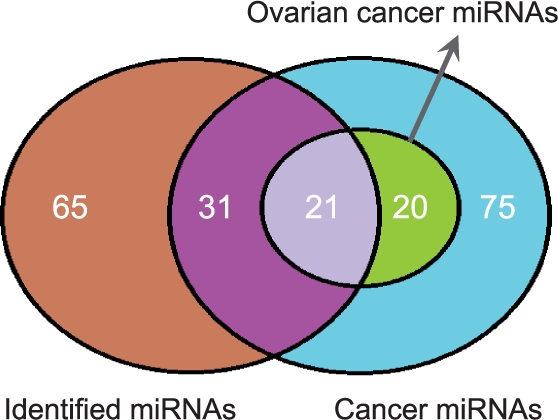
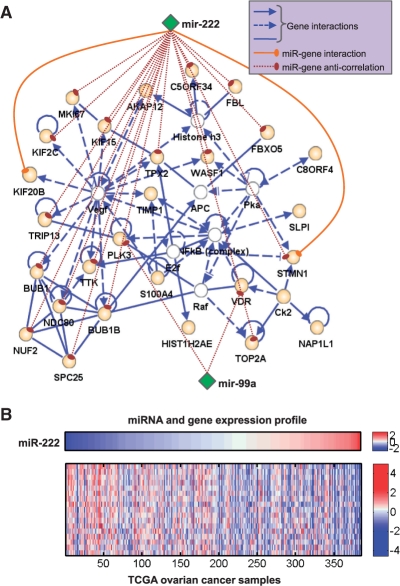
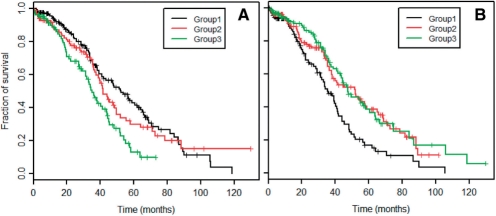
Results: Here we propose an effective data integration framework to identify the miRNA-gene regulatory comodules. The miRNA and gene expression profiles are jointly analyzed in a multiple non-negative matrix factorization framework, and additional network data are simultaneously integrated in a regularized manner. Meanwhile, we employ the sparsity penalties to the variables to achieve modular solutions. The mathematical formulation can be effectively solved by an iterative multiplicative updating algorithm. We apply the proposed method to integrate a set of heterogeneous data sources including the expression profiles of miRNAs and genes on 385 human ovarian cancer samples, computationally predicted miRNA-gene interactions, and gene-gene interactions. We demonstrate that the miRNAs and genes in 69% of the regulatory comodules are significantly associated. Moreover, the comodules are significantly enriched in known functional sets such as miRNA clusters, GO biological processes and KEGG pathways, respectively. Furthermore, many miRNAs and genes in the comodules are related with various cancers including ovarian cancer. Finally, we show that comodules can stratify patients (samples) into groups with significant clinical characteristics.
Availability: The program and supplementary materials are available at http://zhoulab.usc.edu/SNMNMF/.
Contact: xjzhou@usc.edu; zsh@amss.ac.cn
Figures
References
-
- Berry M., et al. Algorithms and applications for approximation nonnegative matrix factorization. Comput. Stat. Data Anal. 2007;52:155–173.
-
- Bartel D.P. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. - PubMed
-
- Bentwich I., et al. Identification of hundreds of conserved and nonconserved human microRNAs. Nat. Genet. 2005;37:766–770. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
