

vipR: variant identification in pooled DNA using R
- PMID: 21685105
- PMCID: PMC3117388
- DOI: 10.1093/bioinformatics/btr205
vipR: variant identification in pooled DNA using R
Abstract
Motivation: High-throughput-sequencing (HTS) technologies are the method of choice for screening the human genome for rare sequence variants causing susceptibility to complex diseases. Unfortunately, preparation of samples for a large number of individuals is still very cost- and labor intensive. Thus, recently, screens for rare sequence variants were carried out in samples of pooled DNA, in which equimolar amounts of DNA from multiple individuals are mixed prior to sequencing with HTS. The resulting sequence data, however, poses a bioinformatics challenge: the discrimination of sequencing errors from real sequence variants present at a low frequency in the DNA pool.
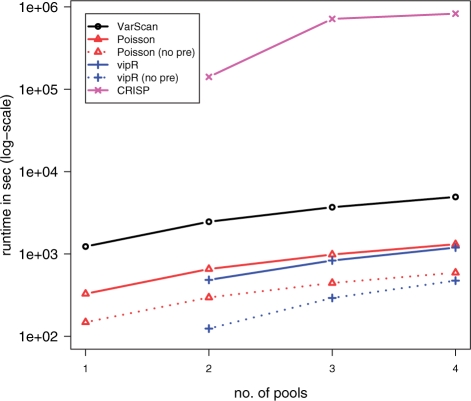
Results: Our method vipR uses data from multiple DNA pools in order to compensate for differences in sequencing error rates along the sequenced region. More precisely, instead of aiming at discriminating sequence variants from sequencing errors, vipR identifies sequence positions that exhibit significantly different minor allele frequencies in at least two DNA pools using the Skellam distribution. The performance of vipR was compared with three other models on data from a targeted resequencing study of the TMEM132D locus in 600 individuals distributed over four DNA pools. Performance of the methods was computed on SNPs that were also genotyped individually using a MALDI-TOF technique. On a set of 82 sequence variants, vipR achieved an average sensitivity of 0.80 at an average specificity of 0.92, thus outperforming the reference methods by at least 0.17 in specificity at comparable sensitivity.
Availability: The code of vipR is freely available via: http://sourceforge.net/projects/htsvipr/
Contact: altmann@mpipsykl.mpg.de.
Figures
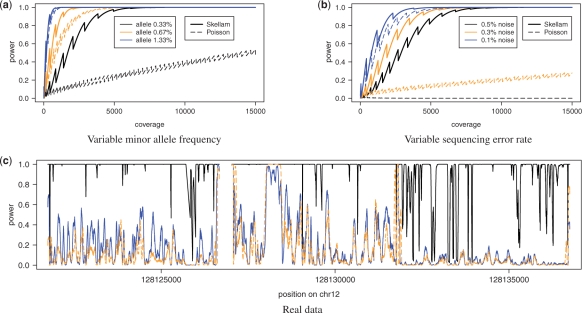
 . (c) Statistical power on real data for the Skellam model (black solid line) using one controls and one cases pool and for the Poisson model separately on one cases (blue solid line) and one controls (orange dashed line) pool.
. (c) Statistical power on real data for the Skellam model (black solid line) using one controls and one cases pool and for the Poisson model separately on one cases (blue solid line) and one controls (orange dashed line) pool.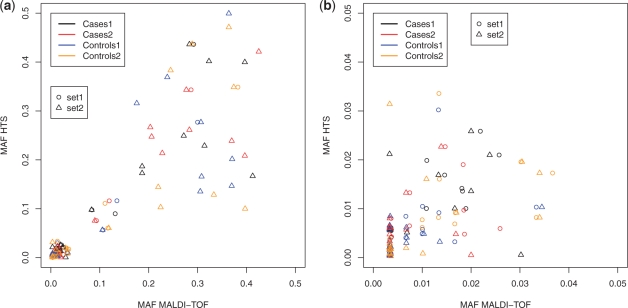
References
-
- Dalca A.V., Brudno M. Genome variation discovery with high-throughput sequencing data. Brief. Bioinformatics. 2010;11:3–14. - PubMed
