

Mass spectrometric identification of novel lysine acetylation sites in huntingtin
- PMID: 21685499
- PMCID: PMC3205870
- DOI: 10.1074/mcp.M111.009829
Mass spectrometric identification of novel lysine acetylation sites in huntingtin
Abstract
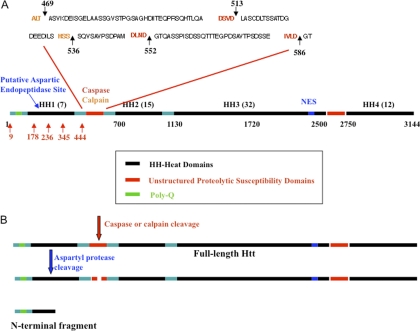
Huntingtin (Htt) is a protein with a polyglutamine stretch in the N-terminus and expansion of the polyglutamine stretch causes Huntington's disease (HD). Htt is a multiple domain protein whose function has not been well characterized. Previous reports have shown, however, that post-translational modifications of Htt such as phosphorylation and acetylation modulate mutant Htt toxicity, localization, and vesicular trafficking. Lysine acetylation of Htt is of particular importance in HD as this modification regulates disease progression and toxicity. Treatment of mouse models with histone deacetylase inhibitors ameliorates HD-like symptoms and alterations in acetylation of Htt promotes clearance of the protein. Given the importance of acetylation in HD and other diseases, we focused on the systematic identification of lysine acetylation sites in Htt23Q (1-612) in a cell culture model using mass spectrometry. Myc-tagged Htt23Q (1-612) overexpressed in the HEK 293T cell line was immunoprecipitated, separated by SDS-PAGE, digested and subjected to high performance liquid chromatography tandem MS analysis. Five lysine acetylation sites were identified, including three novel sites Lys-178, Lys-236, Lys-345 and two previously described sites Lys-9 and Lys-444. Antibodies specific to three of the Htt acetylation sites were produced and confirmed the acetylation sites in Htt. A multiple reaction monitoring MS assay was developed to compare quantitatively the Lys-178 acetylation level between wild-type Htt23Q and mutant Htt148Q (1-612). This report represents the first comprehensive mapping of lysine acetylation sites in N-terminal region of Htt.
Figures





References
-
- The Huntington's Disease Research Group, H. D. C. R. (1993) A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell 72, 971–983 - PubMed
-
- Vonsattel J. P., Myers R. H., Stevens T. J., Ferrante R. J., Bird E. D., Richardson E. P., Jr. (1985) Neuropathological classification of Huntington's disease. J. Neuropathol. Exp. Neurol. 44, 559–577 - PubMed
-
- Zuccato C., Belyaev N., Conforti P., Ooi L., Tartari M., Papadimou E., MacDonald M., Fossale E., Zeitlin S., Buckley N., Cattaneo E. (2007) Widespread disruption of repressor element-1 silencing transcription factor/neuron-restrictive silencer factor occupancy at its target genes in Huntington's disease. J. Neurosci. 27, 6972–6983 - PMC - PubMed
-
- Gafni J., Hermel E., Young J. E., Wellington C. L., Hayden M. R., Ellerby L. M. (2004) Inhibition of calpain cleavage of huntingtin reduces toxicity: accumulation of calpain/caspase fragments in the nucleus. J. Biol. Chem. 279, 20211–20220 - PubMed
-
- Cha J. H. (2000) Transcriptional dysregulation in Huntington's disease. Trends Neurosci. 23, 387–392 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
