

A genetic interaction network of five genes for human polycystic kidney and liver diseases defines polycystin-1 as the central determinant of cyst formation
- PMID: 21685914
- PMCID: PMC3547075
- DOI: 10.1038/ng.860
A genetic interaction network of five genes for human polycystic kidney and liver diseases defines polycystin-1 as the central determinant of cyst formation
Abstract
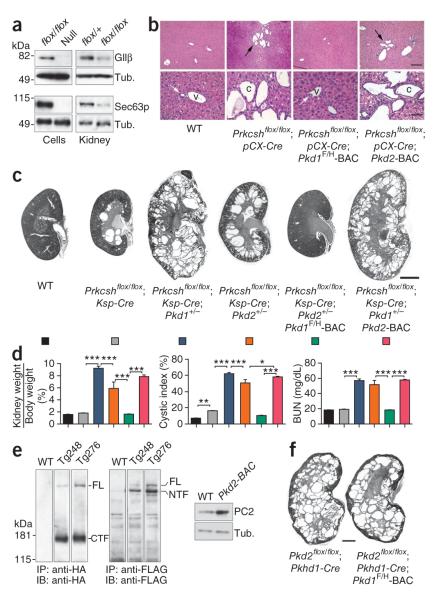
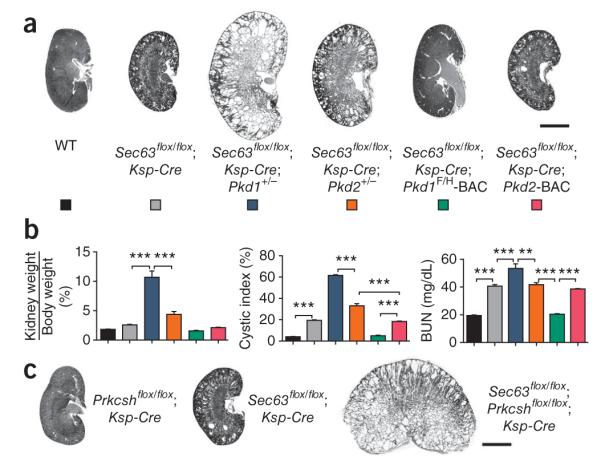
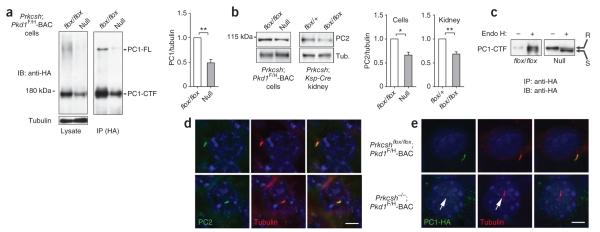
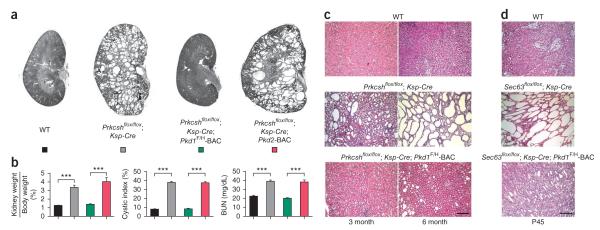
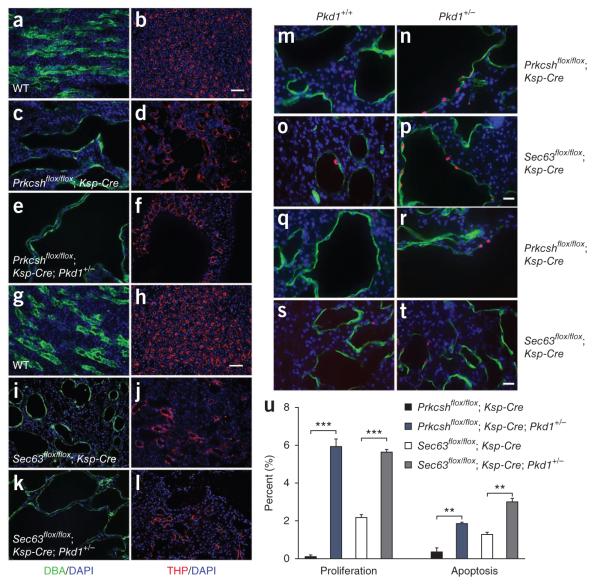
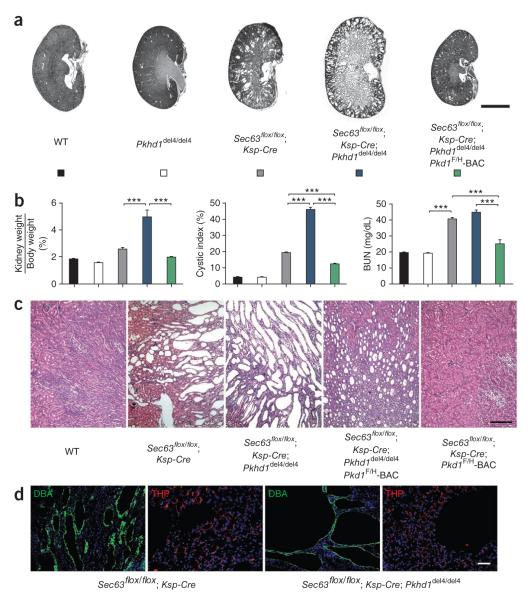
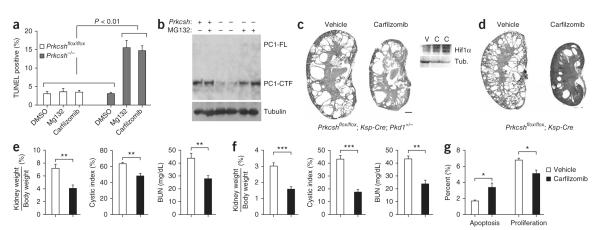
Autosomal dominant polycystic liver disease results from mutations in PRKCSH or SEC63. The respective gene products, glucosidase IIβ and SEC63p, function in protein translocation and quality control pathways in the endoplasmic reticulum. Here we show that glucosidase IIβ and Sec63p are required in mice for adequate expression of a functional complex of the polycystic kidney disease gene products, polycystin-1 and polycystin-2. We find that polycystin-1 is the rate-limiting component of this complex and that there is a dose-response relationship between cystic dilation and levels of functional polycystin-1 following mutation of Prkcsh or Sec63. Reduced expression of polycystin-1 also serves to sensitize the kidney to cyst formation resulting from mutations in Pkhd1, the recessive polycystic kidney disease gene. Finally, we show that proteasome inhibition increases steady-state levels of polycystin-1 in cells lacking glucosidase IIβ and that treatment with a proteasome inhibitor reduces cystic disease in orthologous gene models of human autosomal dominant polycystic liver disease.
Figures
Comment in
-
Polycystin-1: a key player in hereditary cystic kidney and liver disorders.Clin Genet. 2011 Dec;80(6):507-9. doi: 10.1111/j.1399-0004.2011.01780.x. Epub 2011 Sep 23. Clin Genet. 2011. PMID: 21895640 No abstract available.
-
It's not all in the cilium, but on the road to it: genetic interaction network in polycystic kidney and liver diseases and how trafficking and quality control matter.J Hepatol. 2012 May;56(5):1201-1203. doi: 10.1016/j.jhep.2011.10.014. Epub 2011 Nov 28. J Hepatol. 2012. PMID: 22133568
References
-
- Qian Q, et al. Clinical profile of autosomal dominant polycystic liver disease. Hepatology. 2003;37:164–171. - PubMed
-
- Davila S, et al. Mutations in SEC63 cause autosomal dominant polycystic liver disease. Nat. Genet. 2004;36:575–577. - PubMed
-
- Drenth JP, Te Morsche RH, Smink R, Bonifacino JS, Jansen JB. Germline mutations in PRKCSH are associated with autosomal dominant polycystic liver disease. Nat. Genet. 2003;33:345–347. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
- R01DK51041/DK/NIDDK NIH HHS/United States
- R01 DK054053/DK/NIDDK NIH HHS/United States
- P30 DK079310/DK/NIDDK NIH HHS/United States
- R01 AI055914/AI/NIAID NIH HHS/United States
- T32 DK007276/DK/NIDDK NIH HHS/United States
- T32DK007276/DK/NIDDK NIH HHS/United States
- P30DK079310/DK/NIDDK NIH HHS/United States
- R01DK54053/DK/NIDDK NIH HHS/United States
- P50DK57328/DK/NIDDK NIH HHS/United States
- P50 DK057328/DK/NIDDK NIH HHS/United States
- R01 DK051041/DK/NIDDK NIH HHS/United States
- F31DK083904/DK/NIDDK NIH HHS/United States
- F31 DK083904/DK/NIDDK NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
