

T-bet in disease
- PMID: 21685955
- PMCID: PMC6290474
- DOI: 10.1038/ni.2059
T-bet in disease
Abstract
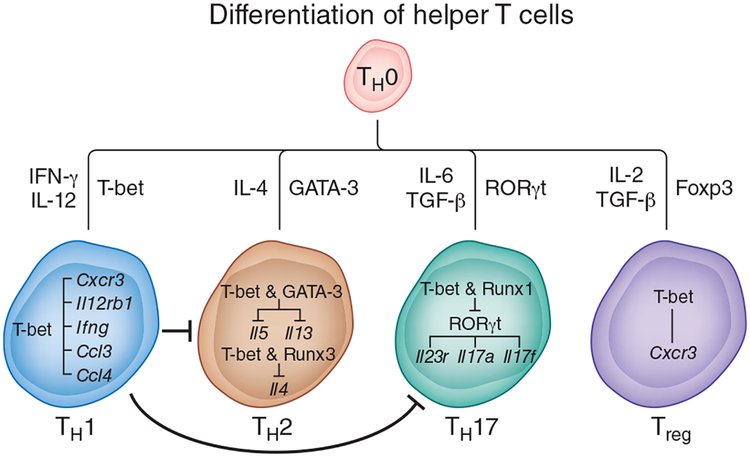
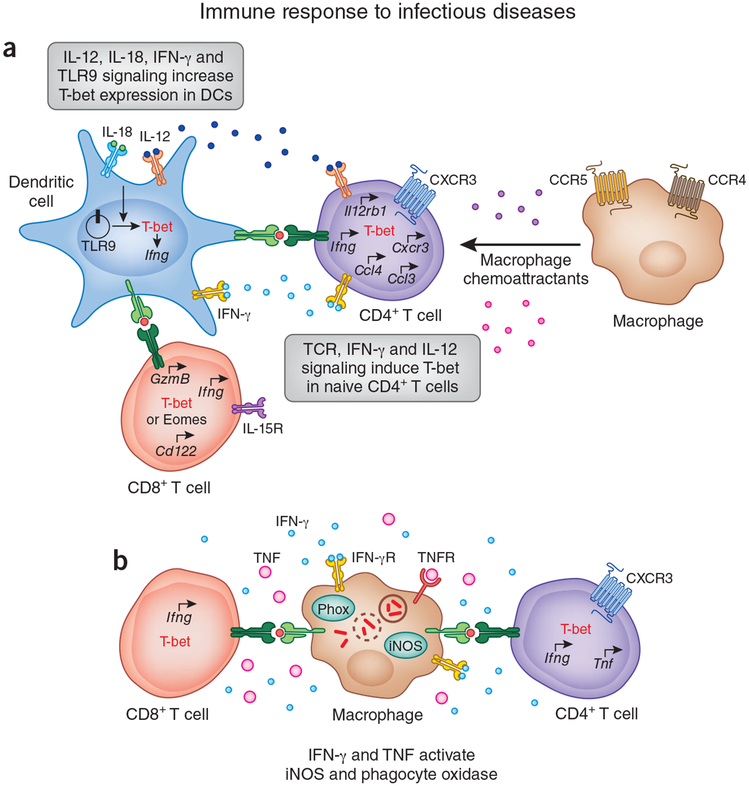
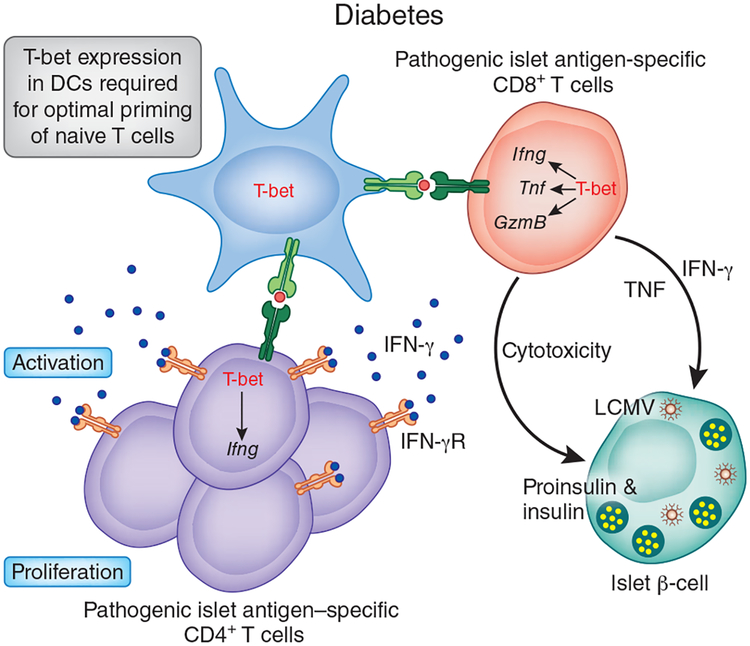
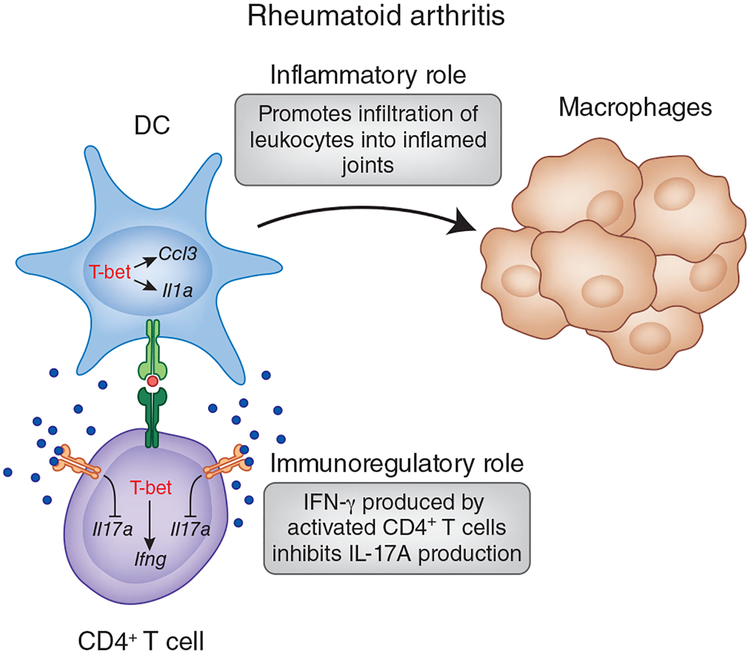
The activation of immune-defense mechanisms in response to a microbial attack must be robust and appropriately tailored to fight particular types of pathogens. Infection with intracellular microorganisms elicits a type 1 inflammatory response characterized by mobilization of T helper type 1 (T(H)1) cells to the site of infection, where they are responsible for the recruitment and activation of macrophages. At the center of the type 1 inflammatory response is the transcription factor T-bet, a critical regulator of the T(H)1 differentiation program. T-bet induces the production of interferon-γ (IFN-γ) and orchestrates the T(H)1 cell-migratory program by regulating the expression of chemokines and chemokine receptors. However, tight regulation of the type 1 inflammatory response is essential for the prevention of immunopathology and the development of organ-specific autoimmunity. In this review, we discuss how T-bet expression drives autoaggressive and inflammatory processes and how its function in vivo must be delicately balanced to avoid disease.
Conflict of interest statement
COMPETING FINANCIAL INTERESTS
The authors declare competing financial interests: details accompany the full-text HTML version of the paper at
Figures
References
-
- Mosmann TR, Cherwinski H, Bond MW, Giedlin MA & Coffman RL Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J. Immunol 136, 2348–2357 (1986). - PubMed
-
- Szabo SJ et al. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell 100, 655–669 (2000). - PubMed
-
- Zheng W & Flavell RA The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell 89, 587–596 (1997). - PubMed
-
- Harrington LE et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat. Immunol 6, 1123–1132 (2005). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
