

Preconditioning involves selective mitophagy mediated by Parkin and p62/SQSTM1
- PMID: 21687634
- PMCID: PMC3110820
- DOI: 10.1371/journal.pone.0020975
Preconditioning involves selective mitophagy mediated by Parkin and p62/SQSTM1
Abstract
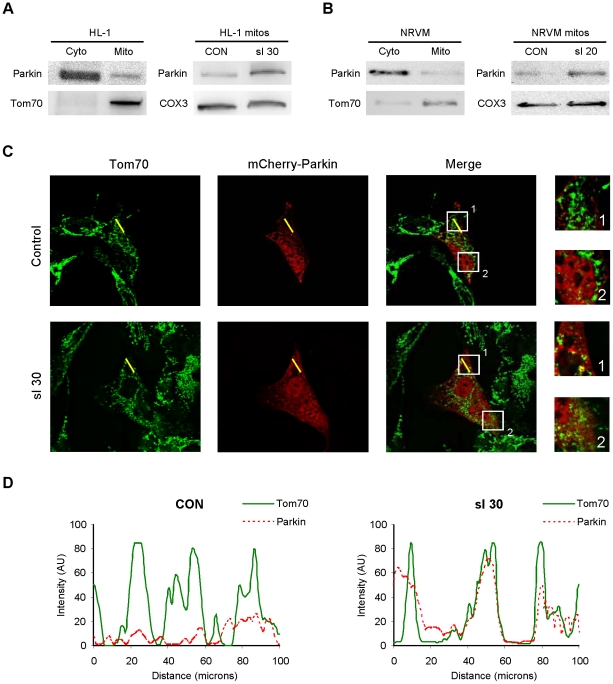
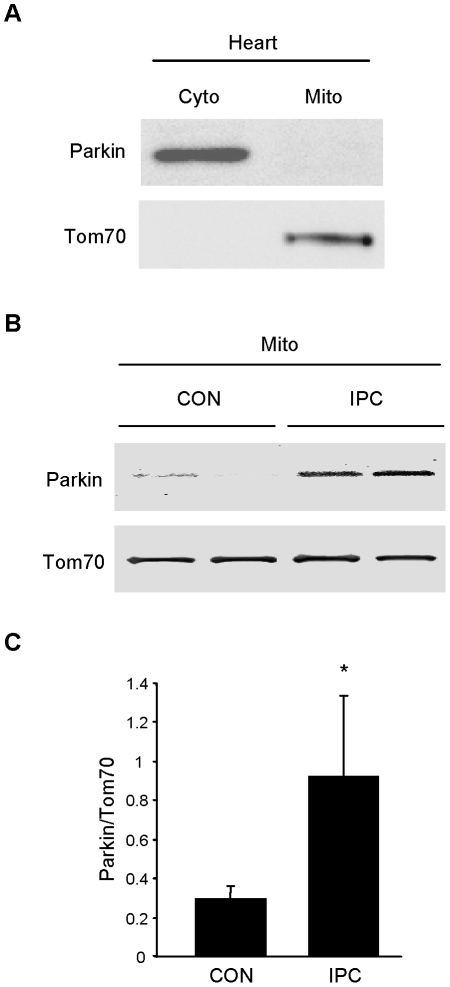
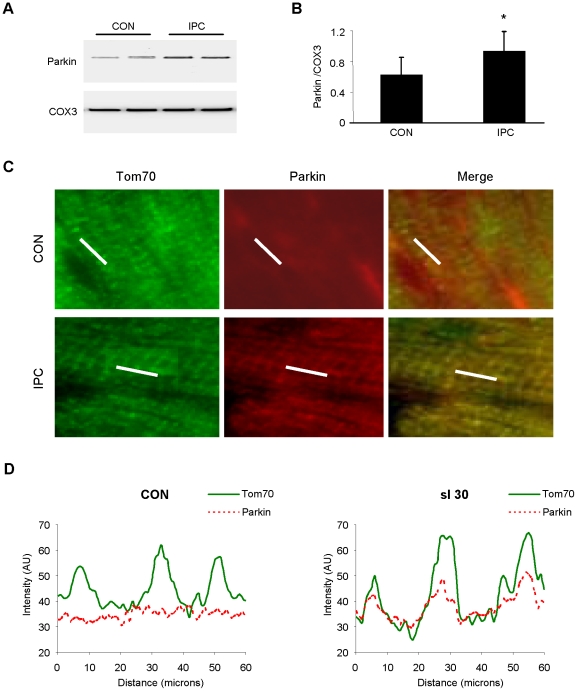
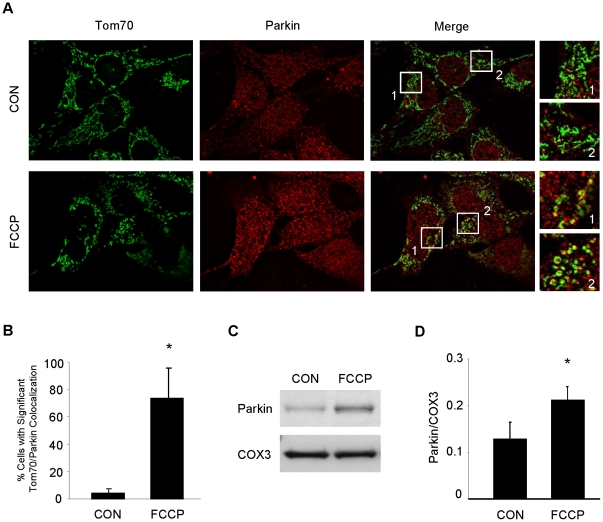
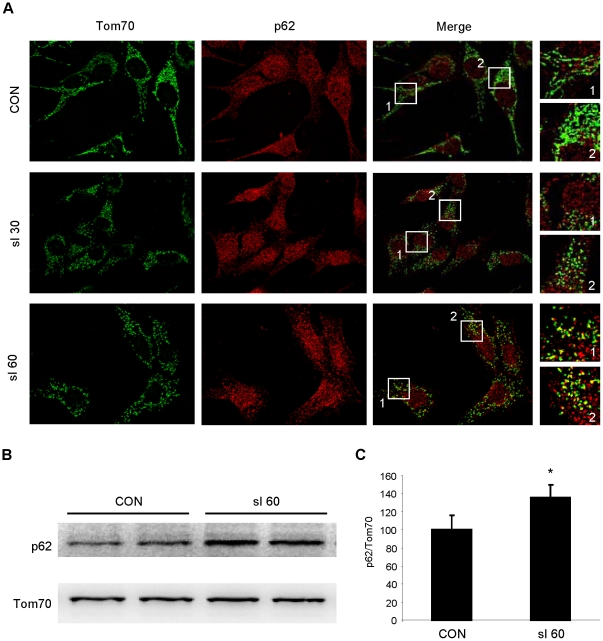
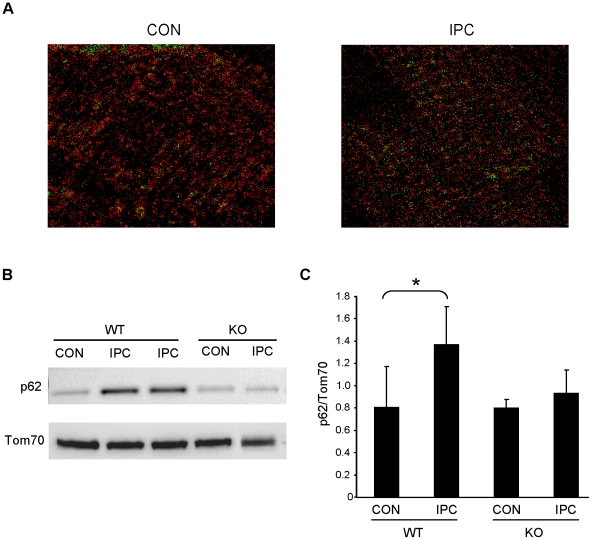
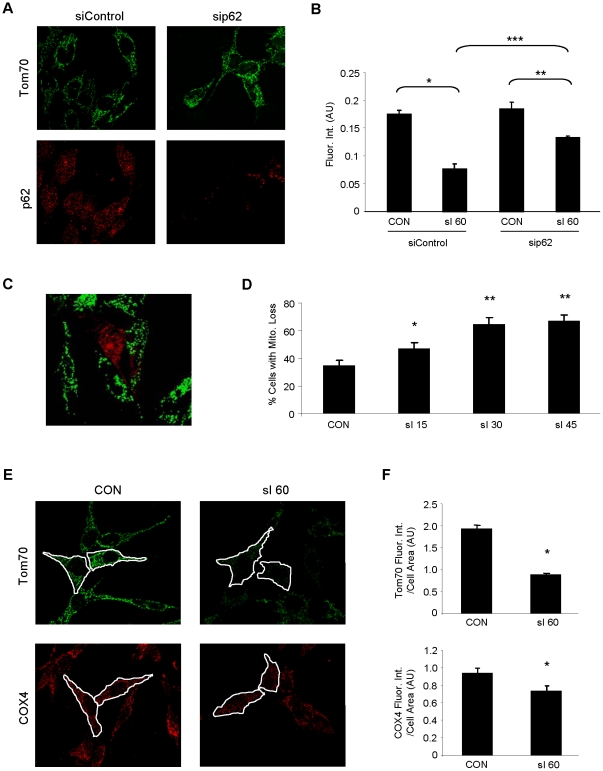
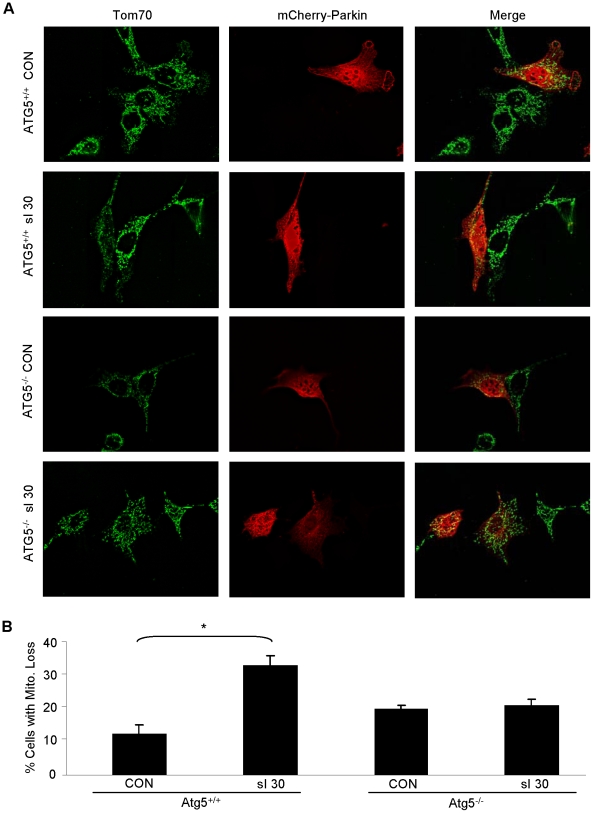
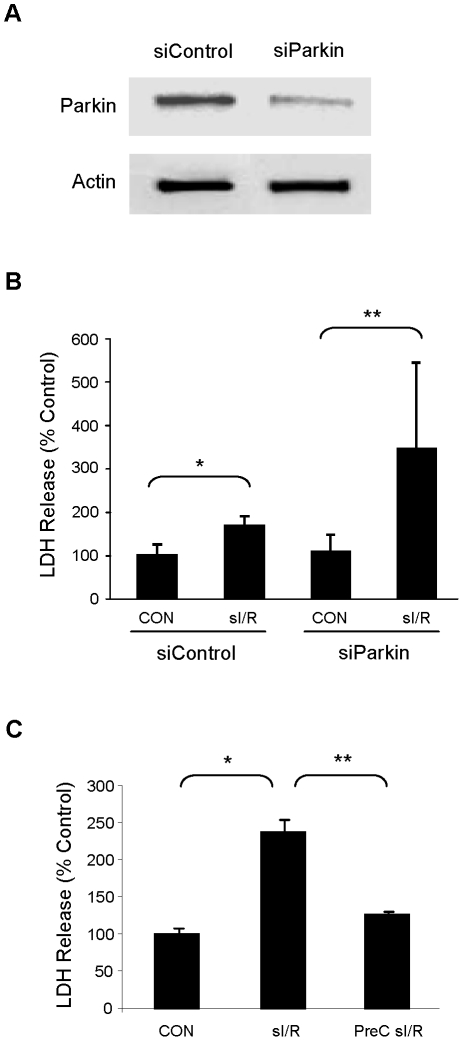
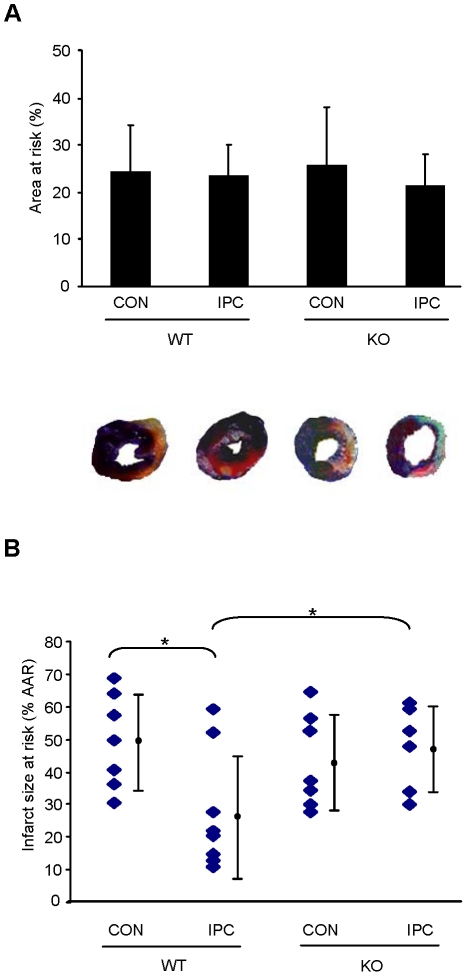
Autophagy-dependent mitochondrial turnover in response to cellular stress is necessary for maintaining cellular homeostasis. However, the mechanisms that govern the selective targeting of damaged mitochondria are poorly understood. Parkin, an E3 ubiquitin ligase, has been shown to be essential for the selective clearance of damaged mitochondria. Parkin is expressed in the heart, yet its function has not been investigated in the context of cardioprotection. We previously reported that autophagy is required for cardioprotection by ischemic preconditioning (IPC). In the present study, we used simulated ischemia (sI) in vitro and IPC of hearts to investigate the role of Parkin in mediating cardioprotection ex vivo and in vivo. In HL-1 cells, sI induced Parkin translocation to mitochondria and mitochondrial elimination. IPC induced Parkin translocation to mitochondria in Langendorff-perfused rat hearts and in vivo in mice subjected to regional IPC. Mitochondrial depolarization with an uncoupling agent similarly induced Parkin translocation to mitochondria in cells and Langendorff-perfused rat hearts. Mitochondrial loss was blunted in Atg5-deficient cells, revealing the requirement for autophagy in mitochondrial elimination. Consistent with previous reports indicating a role for p62/SQSTM1 in mitophagy, we found that depletion of p62 attenuated mitophagy and exacerbated cell death in HL-1 cardiomyocytes subjected to sI. While wild type mice showed p62 translocation to mitochondria and an increase in ubiquitination, Parkin knockout mice exhibited attenuated IPC-induced p62 translocation to the mitochondria. Importantly, ablation of Parkin in mice abolished the cardioprotective effects of IPC. These results reveal for the first time the crucial role of Parkin and mitophagy in cardioprotection.
Conflict of interest statement
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
