Computational methods for Gene Orthology inference
- PMID: 21690100
- PMCID: PMC3178053
- DOI: 10.1093/bib/bbr030
Computational methods for Gene Orthology inference
Abstract
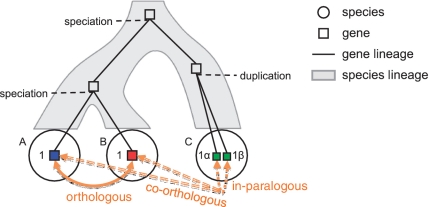
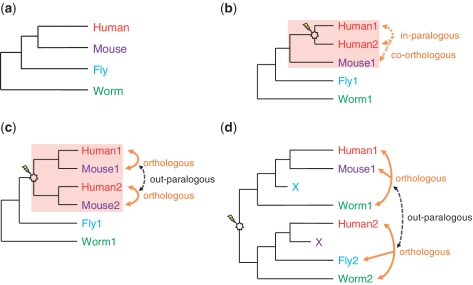
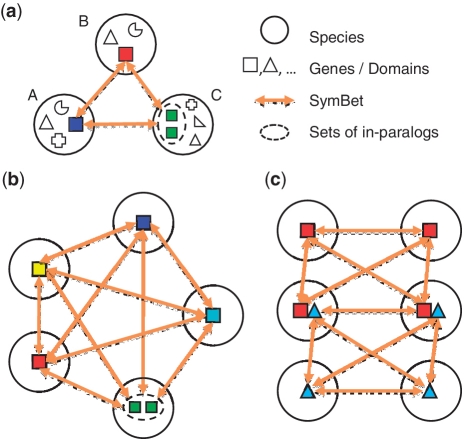
Accurate inference of orthologous genes is a pre-requisite for most comparative genomics studies, and is also important for functional annotation of new genomes. Identification of orthologous gene sets typically involves phylogenetic tree analysis, heuristic algorithms based on sequence conservation, synteny analysis, or some combination of these approaches. The most direct tree-based methods typically rely on the comparison of an individual gene tree with a species tree. Once the two trees are accurately constructed, orthologs are straightforwardly identified by the definition of orthology as those homologs that are related by speciation, rather than gene duplication, at their most recent point of origin. Although ideal for the purpose of orthology identification in principle, phylogenetic trees are computationally expensive to construct for large numbers of genes and genomes, and they often contain errors, especially at large evolutionary distances. Moreover, in many organisms, in particular prokaryotes and viruses, evolution does not appear to have followed a simple 'tree-like' mode, which makes conventional tree reconciliation inapplicable. Other, heuristic methods identify probable orthologs as the closest homologous pairs or groups of genes in a set of organisms. These approaches are faster and easier to automate than tree-based methods, with efficient implementations provided by graph-theoretical algorithms enabling comparisons of thousands of genomes. Comparisons of these two approaches show that, despite conceptual differences, they produce similar sets of orthologs, especially at short evolutionary distances. Synteny also can aid in identification of orthologs. Often, tree-based, sequence similarity- and synteny-based approaches can be combined into flexible hybrid methods.
Figures