

Duchenne muscular dystrophy gene therapy: Lost in translation?
- PMID: 21691429
- PMCID: PMC3117615
- DOI: 10.2147/RRB.S13463
Duchenne muscular dystrophy gene therapy: Lost in translation?
Abstract
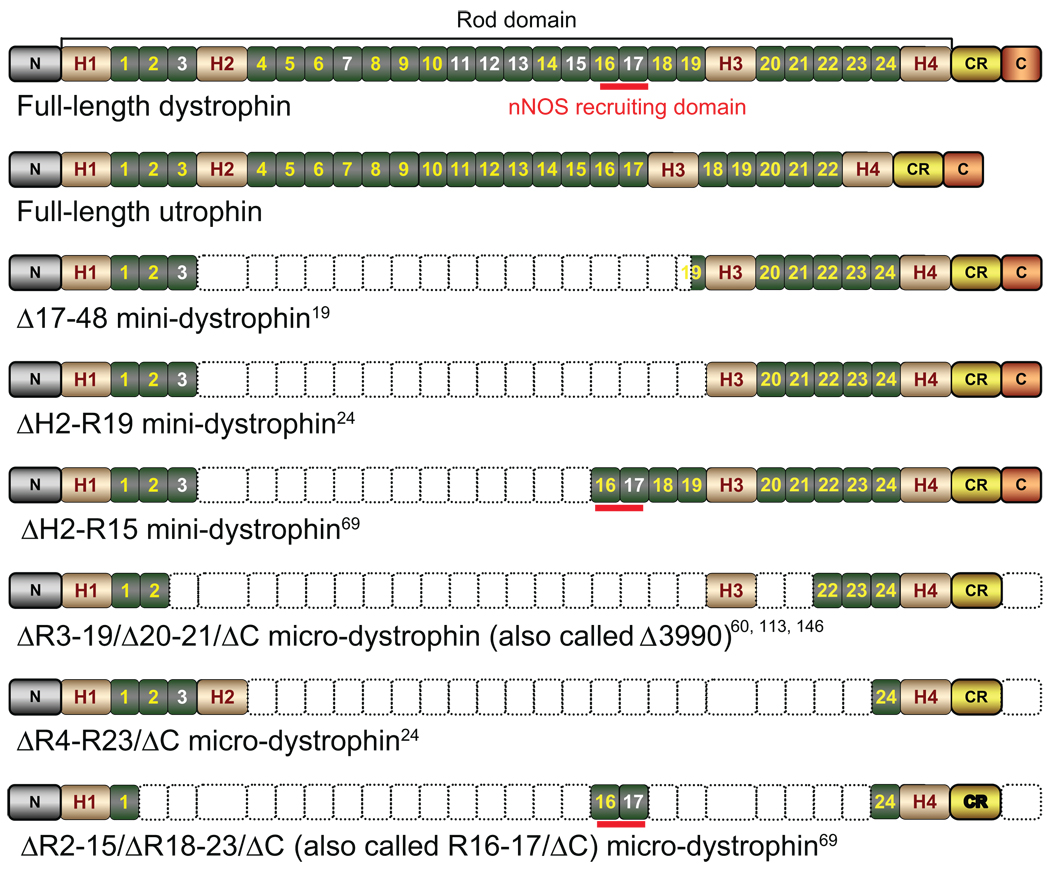
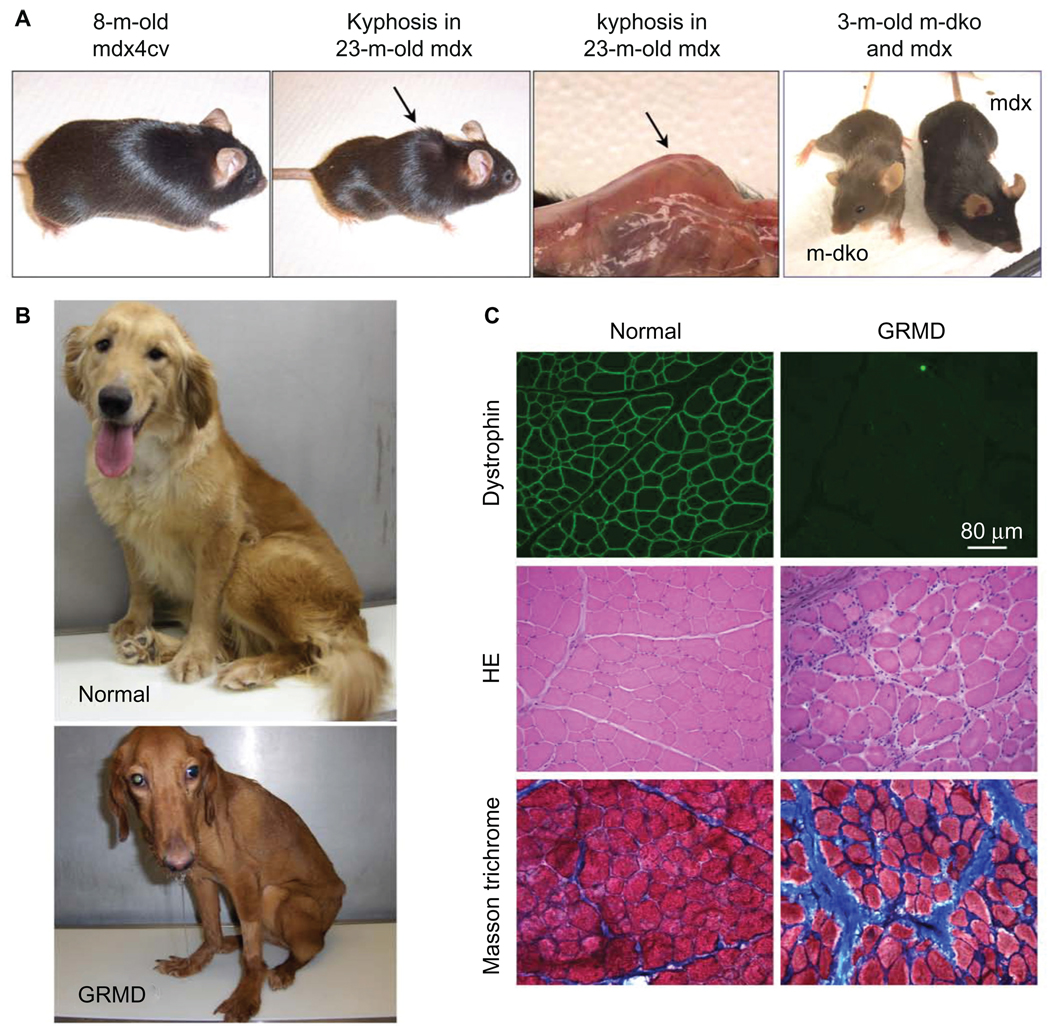
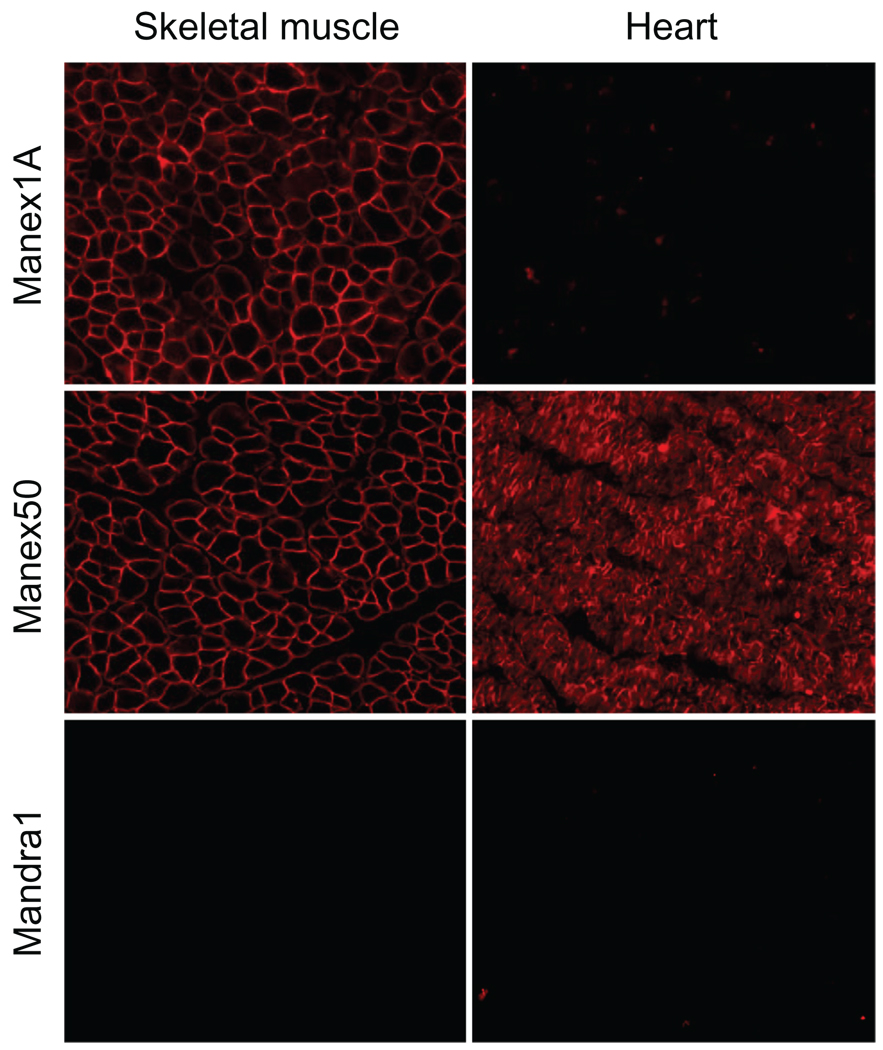
A milestone of molecular medicine is the identification of dystrophin gene mutation as the cause of Duchenne muscular dystrophy (DMD). Over the last 2 decades, major advances in dystrophin biology and gene delivery technology have created an opportunity to treat DMD with gene therapy. Remarkable success has been achieved in treating dystrophic mice. Several gene therapy strategies, including plasmid transfer, exon skipping, and adeno-associated virus-mediated microdystrophin therapy, have entered clinical trials. However, therapeutic benefit has not been realized in DMD patients. Bridging the gap between mice and humans is no doubt the most pressing issue facing DMD gene therapy now. In contrast to mice, dystrophin-deficient dogs are genetically and phenotypically similar to human patients. Preliminary gene therapy studies in the canine model may offer critical insights that cannot be obtained from murine studies. It is clear that the canine DMD model may represent an important link between mice and humans. Unfortunately, our current knowledge of dystrophic dogs is limited, and the full picture of disease progression remains to be clearly defined. We also lack rigorous outcome measures (such as in situ force measurement) to monitor therapeutic efficacy in dystrophic dogs. Undoubtedly, maintaining a dystrophic dog colony is technically demanding, and the cost of dog studies cannot be underestimated. A carefully coordinated effort from the entire DMD community is needed to make the best use of the precious dog resource. Successful DMD gene therapy may depend on valid translational studies in dystrophin-deficient dogs.
Conflict of interest statement
The author reports no conflicts of interest in this work.
Figures
References
-
- Hoffman EP, Brown RH, Jr, Kunkel LM. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51(6):919–928. - PubMed
-
- Koenig M, Hoffman EP, Bertelson CJ, Monaco AP, Feener C, Kunkel LM. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell. 1987;50(3):509–517. - PubMed
-
- Romitti P, Puzhankara S, Mathews K, et al. Centers for Disease Control and Prevention (CDC). Prevalence of Duchenne/Becker muscular dystrophy among males aged 5–24 years – four states, 2007. Morb Mortal Wkly Rep. 2009;58(40):1119–1122. - PubMed
-
- Dooley J, Gordon KE, Dodds L, MacSween J. Duchenne muscular dystrophy: a 30-year population-based incidence study. Clin Pediatr (Phila) 2010;49(2):177–179. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous