

Congenital hepatic fibrosis and portal hypertension in autosomal dominant polycystic kidney disease
- PMID: 21694639
- PMCID: PMC8366680
- DOI: 10.1097/MPG.0b013e318228330c
Congenital hepatic fibrosis and portal hypertension in autosomal dominant polycystic kidney disease
Abstract
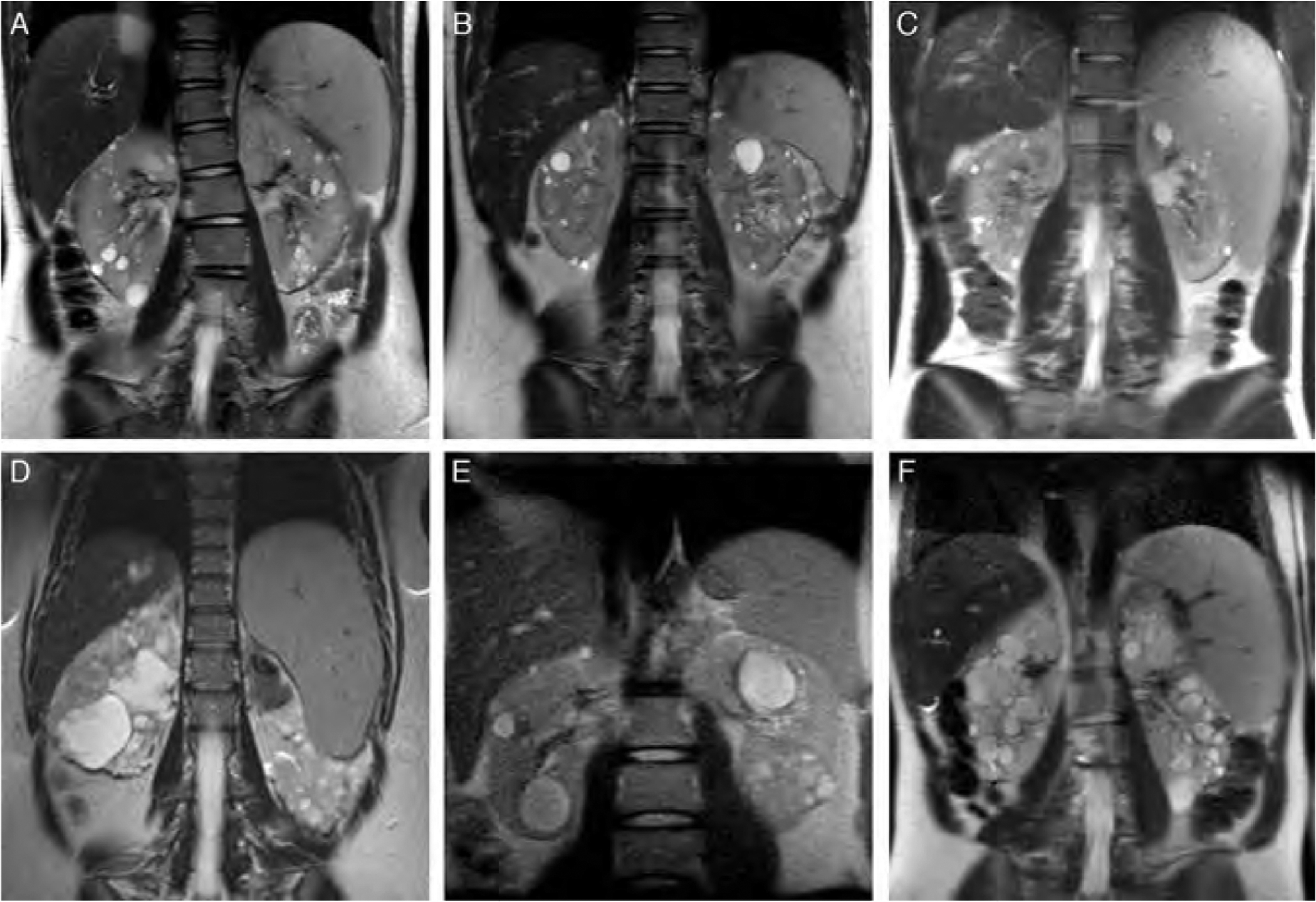
Objectives: Autosomal dominant (ADPKD) and recessive (ARPKD) polycystic kidney diseases are the most common hepatorenal fibrocystic diseases (ciliopathies). Characteristics of liver disease of these disorders are quite different. All of the patients with ARPKD have congenital hepatic fibrosis (CHF) often complicated by portal hypertension. In contrast, typical liver involvement in ADPKD is polycystic liver disease, although rare atypical cases with CHF are reported. Our goal was to describe the characteristics of CHF in ADPKD.
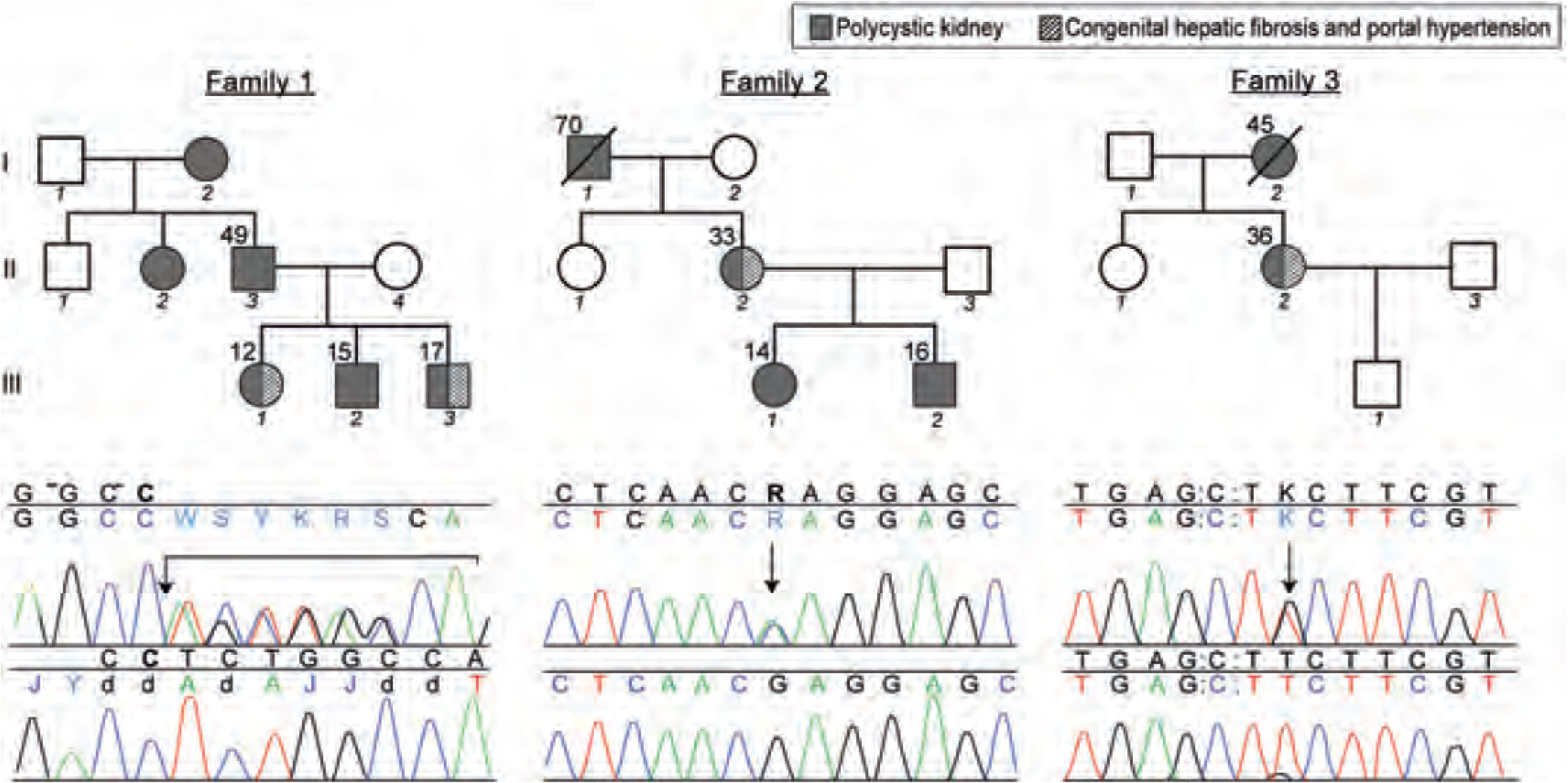
Patients and methods: As a part of an intramural study of the National Institutes of Health on ciliopathies (www.clinicaltrials.gov, trial NCT00068224), we evaluated 8 patients from 3 ADPKD families with CHF. We present their clinical, biochemical, imaging, and PKD1 and PKHD1 sequencing results. In addition, we tabulate the characteristics of 15 previously reported patients with ADPKD-CHF from 11 families.
Results: In all of the 19 patients with ADPKD-CHF (9 boys, 10 girls), portal hypertension was the main manifestation of CHF; hepatocelllular function was preserved and liver enzymes were largely normal. In all of the 14 families, CHF was not inherited vertically, that is the parents of the index cases had PKD but did not have CHF-suggesting modifier gene(s). Our 3 families had pathogenic mutations in PKD1; sequencing of the PKHD1 gene as a potential modifier did not reveal any mutations.
Conclusions: Characteristics of CHF in ADPKD are similar to CHF in ARPKD. ADPKD-CHF is caused by PKD1 mutations, with probable contribution from modifying gene(s). Given that both boys and girls are affected, these modifier(s) are likely located on autosomal chromosome(s) and less likely X-linked.
Conflict of interest statement
The authors report no conflicts of interest.
Figures
References
-
- Harris PC, Torres VE. Polycystic kidney disease, autosomal dominant. In: GeneReviews at GeneTests: Medical Genetics Information Resource. University of Washington, Seattle: 1997–2008. Available at http://www.genetests.org.AccessedNovember 10, 2010. - PubMed
-
- Arnold HL, Harrison SA. New advances in evaluation and management of patients with polycystic liver disease. Am J Gastroenterol 2005;100:2569–82. - PubMed
-
- Hughes J, Ward CJ, Peral B, et al.The polycystic kidney disease 1 (PKD1) gene encodes a novel protein with multiple cell recognition domains. Nat Genet 1995;10:151–60. - PubMed
-
- Mochizuki T, Wu G, Hayashi T, et al.PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science 1996;272:1339–42. - PubMed
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
