

DADA: Degree-Aware Algorithms for Network-Based Disease Gene Prioritization
- PMID: 21699738
- PMCID: PMC3143097
- DOI: 10.1186/1756-0381-4-19
DADA: Degree-Aware Algorithms for Network-Based Disease Gene Prioritization
Abstract
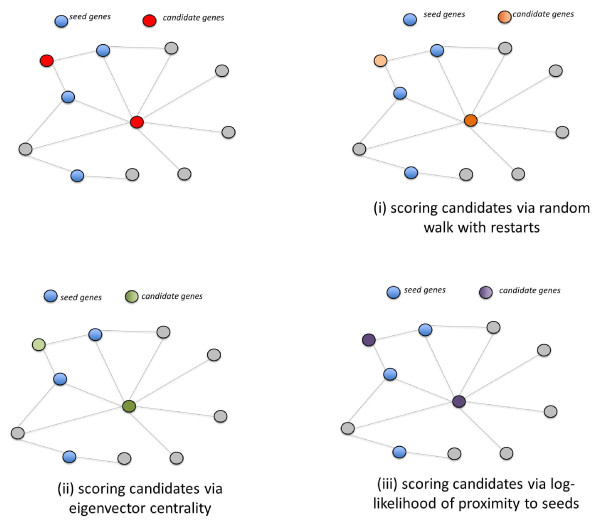
Background: High-throughput molecular interaction data have been used effectively to prioritize candidate genes that are linked to a disease, based on the observation that the products of genes associated with similar diseases are likely to interact with each other heavily in a network of protein-protein interactions (PPIs). An important challenge for these applications, however, is the incomplete and noisy nature of PPI data. Information flow based methods alleviate these problems to a certain extent, by considering indirect interactions and multiplicity of paths.
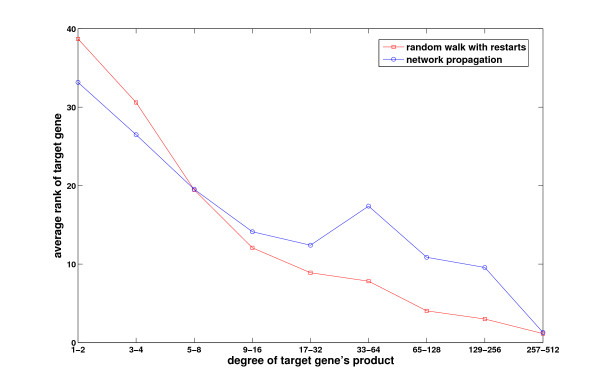
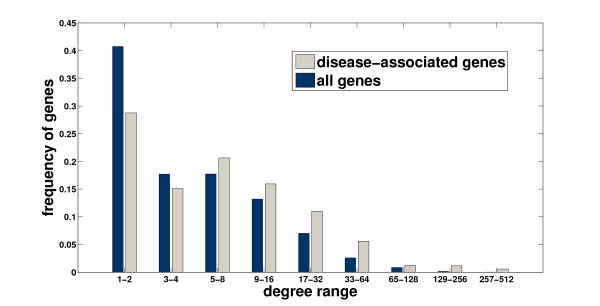
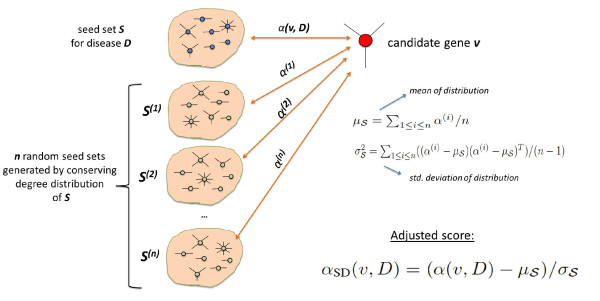
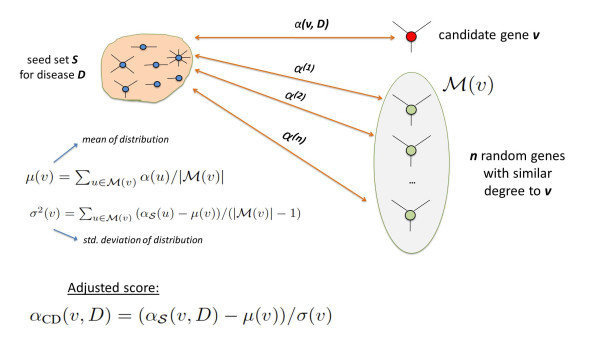
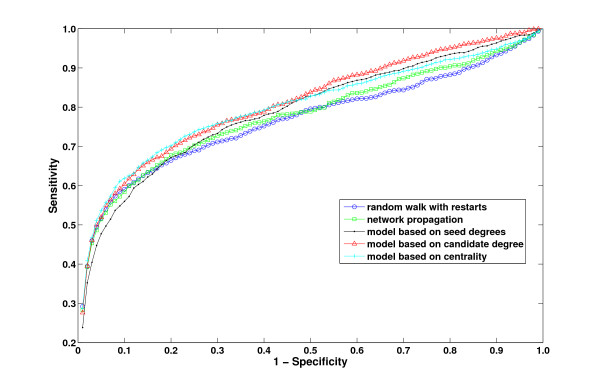
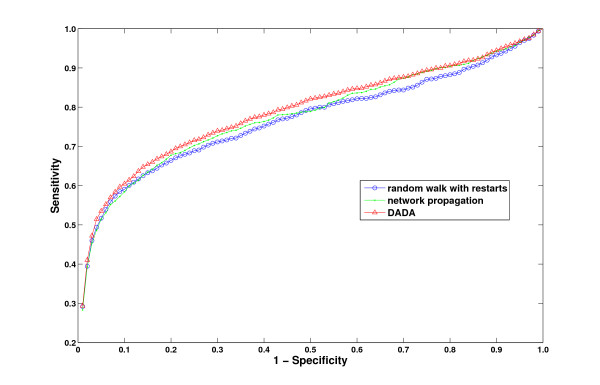
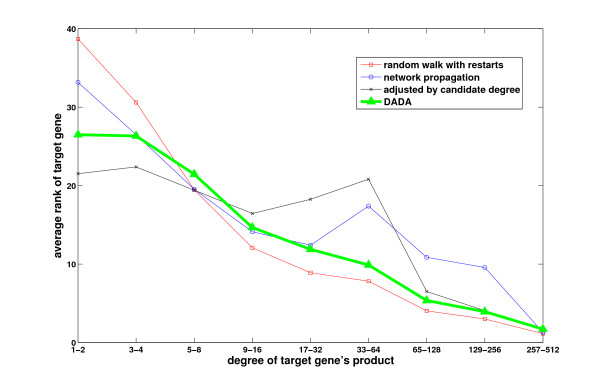
Results: We demonstrate that existing methods are likely to favor highly connected genes, making prioritization sensitive to the skewed degree distribution of PPI networks, as well as ascertainment bias in available interaction and disease association data. Motivated by this observation, we propose several statistical adjustment methods to account for the degree distribution of known disease and candidate genes, using a PPI network with associated confidence scores for interactions. We show that the proposed methods can detect loosely connected disease genes that are missed by existing approaches, however, this improvement might come at the price of more false negatives for highly connected genes. Consequently, we develop a suite called DADA, which includes different uniform prioritization methods that effectively integrate existing approaches with the proposed statistical adjustment strategies. Comprehensive experimental results on the Online Mendelian Inheritance in Man (OMIM) database show that DADA outperforms existing methods in prioritizing candidate disease genes.
Conclusions: These results demonstrate the importance of employing accurate statistical models and associated adjustment methods in network-based disease gene prioritization, as well as other network-based functional inference applications. DADA is implemented in Matlab and is freely available at http://compbio.case.edu/dada/.
Figures

References
-
- Brunner HG, van Driel MA. From syndrome families to functional genomics. Nat Rev Genet. 2004;5(7):545–551. - PubMed
-
- Glazier AM, Nadeau JH, Aitman TJ. Finding Genes That Underlie Complex Traits. Science. 2002;298(5602):2345–2349. doi: 10.1126/science.1076641. http://www.sciencemag.org/cgi/content/abstract/298/5602/2345 - DOI - PubMed
-
- Adie E, Adams R, Evans K, Porteous D, Pickard B. SUSPECTS: enabling fast and effective prioritization of positional candidates. Bioinformatics. 2006;22(6):773–774. doi: 10.1093/bioinformatics/btk031. http://bioinformatics.oxfordjournals.org/cgi/content/abstract/22/6/773 - DOI - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
