

Using VAAST to identify an X-linked disorder resulting in lethality in male infants due to N-terminal acetyltransferase deficiency
- PMID: 21700266
- PMCID: PMC3135802
- DOI: 10.1016/j.ajhg.2011.05.017
Using VAAST to identify an X-linked disorder resulting in lethality in male infants due to N-terminal acetyltransferase deficiency
Erratum in
- Am J Hum Genet. 2011 Aug 12;89(2):345
Abstract
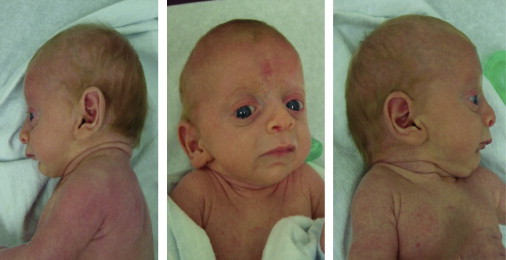
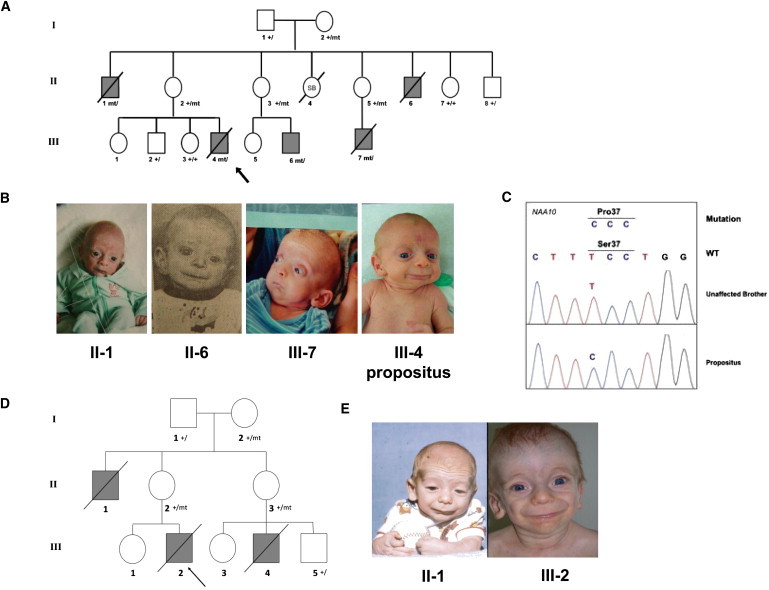
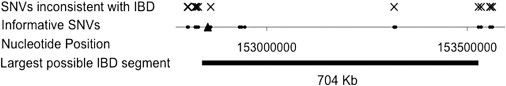
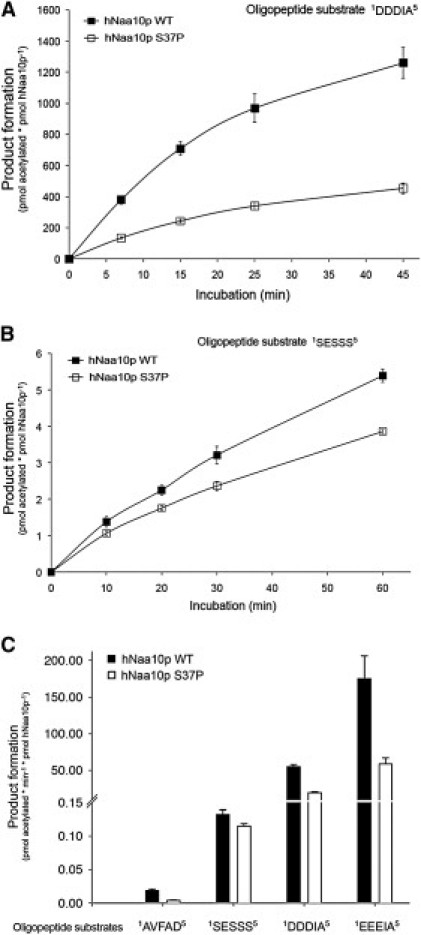
We have identified two families with a previously undescribed lethal X-linked disorder of infancy; the disorder comprises a distinct combination of an aged appearance, craniofacial anomalies, hypotonia, global developmental delays, cryptorchidism, and cardiac arrhythmias. Using X chromosome exon sequencing and a recently developed probabilistic algorithm aimed at discovering disease-causing variants, we identified in one family a c.109T>C (p.Ser37Pro) variant in NAA10, a gene encoding the catalytic subunit of the major human N-terminal acetyltransferase (NAT). A parallel effort on a second unrelated family converged on the same variant. The absence of this variant in controls, the amino acid conservation of this region of the protein, the predicted disruptive change, and the co-occurrence in two unrelated families with the same rare disorder suggest that this is the pathogenic mutation. We confirmed this by demonstrating a significantly impaired biochemical activity of the mutant hNaa10p, and from this we conclude that a reduction in acetylation by hNaa10p causes this disease. Here we provide evidence of a human genetic disorder resulting from direct impairment of N-terminal acetylation, one of the most common protein modifications in humans.
Copyright © 2011 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
- RC2 HG005619/HG/NHGRI NIH HHS/United States
- 1RC2HG005619/HG/NHGRI NIH HHS/United States
- UL1 RR025764/RR/NCRR NIH HHS/United States
- UL1RR025764/RR/NCRR NIH HHS/United States
- P30CA042014/CA/NCI NIH HHS/United States
- UL1 RR025774/RR/NCRR NIH HHS/United States
- R01 HG005692/HG/NHGRI NIH HHS/United States
- R01 GM104390/GM/NIGMS NIH HHS/United States
- R44 HG006579/HG/NHGRI NIH HHS/United States
- K99 HG005846/HG/NHGRI NIH HHS/United States
- 5R01HG5692/HG/NHGRI NIH HHS/United States
- K99HG005846/HG/NHGRI NIH HHS/United States
- P30 CA042014/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
