

Activated AMPK inhibits PPAR-{alpha} and PPAR-{gamma} transcriptional activity in hepatoma cells
- PMID: 21700905
- PMCID: PMC3191559
- DOI: 10.1152/ajpgi.00432.2010
Activated AMPK inhibits PPAR-{alpha} and PPAR-{gamma} transcriptional activity in hepatoma cells
Abstract
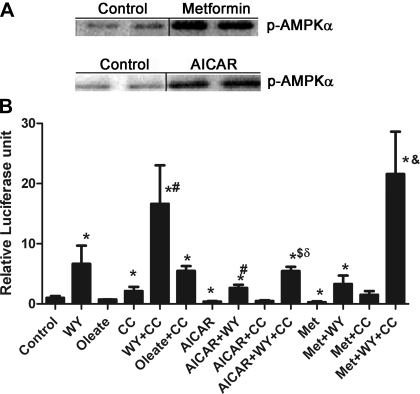
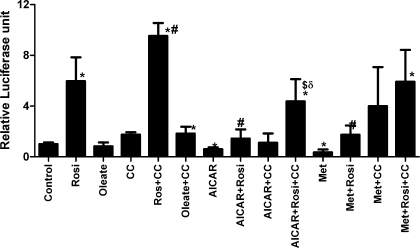
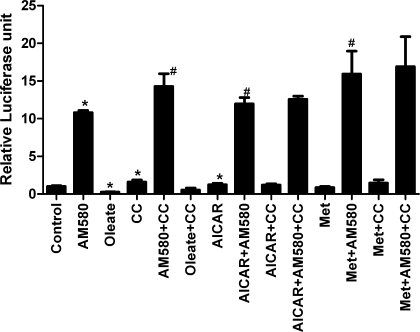
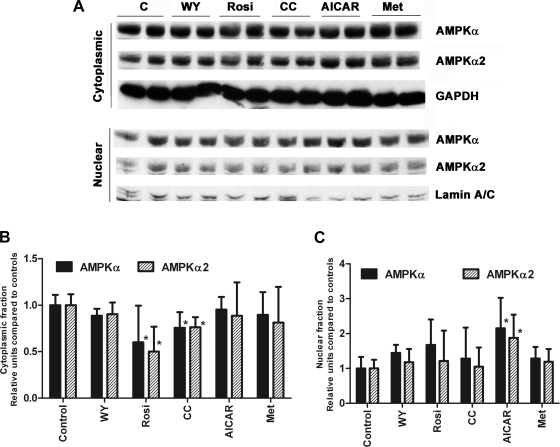
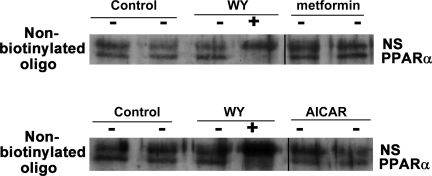
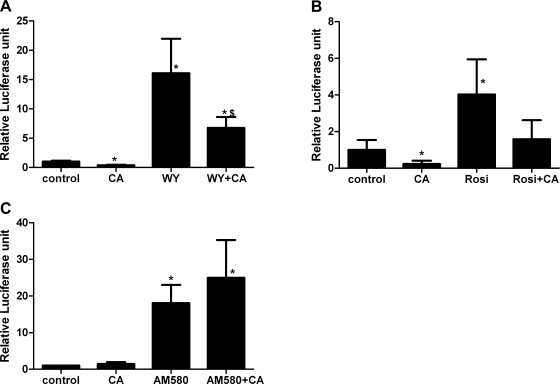
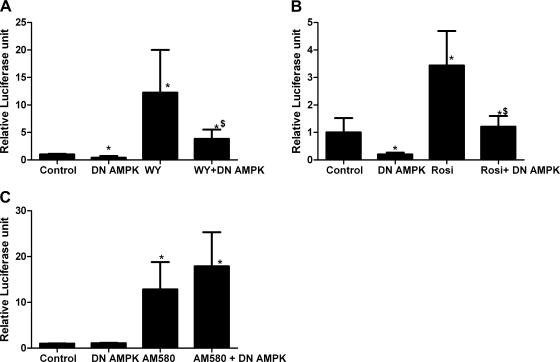
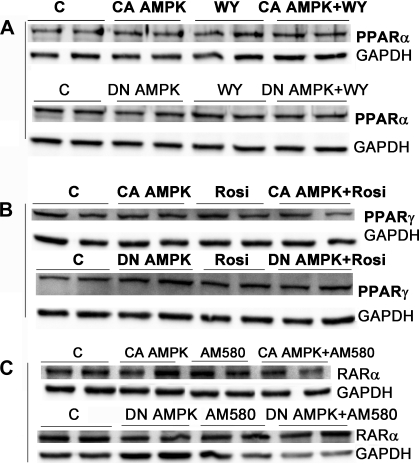
AMP-activated protein kinase (AMPK) and peroxisome proliferator-activated receptor-α (PPAR-α) are critical regulators of short-term and long-term fatty acid oxidation, respectively. We examined whether the activities of these molecules were coordinately regulated. H4IIEC3 cells were transfected with PPAR-α and PPAR-γ expression plasmids and a peroxisome-proliferator-response element (PPRE) luciferase reporter plasmid. The cells were treated with PPAR agonists (WY-14,643 and rosiglitazone), AMPK activators 5-aminoimidazole-4-carboxamide riboside (AICAR) and metformin, and the AMPK inhibitor compound C. Both AICAR and metformin decreased basal and WY-14,643-stimulated PPAR-α activity; compound C increased agonist-stimulated reporter activity and partially reversed the effect of the AMPK activators. Similar effects on PPAR-γ were seen, with both AICAR and metformin inhibiting PPRE reporter activity. Compound C increased basal PPAR-γ activity and rosiglitazone-stimulated activity. In contrast, retinoic acid receptor-α (RAR-α), another nuclear receptor that dimerizes with retinoid X receptor (RXR), was largely unaffected by the AMPK activators. Compound C modestly increased AM580 (an RAR agonist)-stimulated activity. The AMPK activators did not affect PPAR-α binding to DNA, and there was no consistent correlation between effects of the AMPK activators and inhibitor on PPAR and the nuclear localization of AMPK-α subunits. Expression of either a constitutively active or dominant negative AMPK-α inhibited basal and WY-14,643-stimulated PPAR-α activity and basal and rosiglitazone-stimulated PPAR-γ activity. We concluded that the AMPK activators AICAR and metformin inhibited transcriptional activities of PPAR-α and PPAR-γ, whereas inhibition of AMPK with compound C activated both PPARs. The effects of AMPK do not appear to be mediated through effects on RXR or on PPAR/RXR binding to DNA. These effects are independent of kinase activity and instead appear to rely on the activated conformation of AMPK. AMPK inhibition of PPAR-α and -γ may allow for short-term processes to increase energy generation before the cells devote resources to increasing their capacity for fatty acid oxidation.
Figures
Similar articles
-
Anti-inflammatory effects of thiazolidinediones in human airway smooth muscle cells.Am J Respir Cell Mol Biol. 2011 Jul;45(1):111-9. doi: 10.1165/rcmb.2009-0445OC. Epub 2010 Sep 24. Am J Respir Cell Mol Biol. 2011. PMID: 20870897 Free PMC article.
-
Suppression effects of AICAR on insulin secretion involved in peroxisome proliferator-activated receptor gamma changes in INS-1 cells.J Endocrinol Invest. 2010 Jul-Aug;33(7):465-71. doi: 10.1007/BF03346626. Epub 2010 Jan 22. J Endocrinol Invest. 2010. PMID: 20101096
-
AICAR and metformin, but not exercise, increase muscle glucose transport through AMPK-, ERK-, and PDK1-dependent activation of atypical PKC.Am J Physiol Endocrinol Metab. 2010 Feb;298(2):E179-92. doi: 10.1152/ajpendo.00392.2009. Epub 2009 Nov 3. Am J Physiol Endocrinol Metab. 2010. PMID: 19887597 Free PMC article.
-
Regulation of energy metabolism by long-chain fatty acids.Prog Lipid Res. 2014 Jan;53:124-44. doi: 10.1016/j.plipres.2013.12.001. Epub 2013 Dec 18. Prog Lipid Res. 2014. PMID: 24362249 Review.
-
Highlighting the Protective or Degenerative Role of AMPK Activators in Dementia Experimental Models.CNS Neurol Disord Drug Targets. 2021;20(9):786-801. doi: 10.2174/1871527320666210526160214. CNS Neurol Disord Drug Targets. 2021. PMID: 34042039 Review.
Cited by
-
Doxorubicin Attenuates Free Fatty Acid-Induced Lipid Accumulation via Stimulation of p53 in HepG2 Cells.Biomol Ther (Seoul). 2024 Jan 1;32(1):94-103. doi: 10.4062/biomolther.2023.200. Biomol Ther (Seoul). 2024. PMID: 38148555 Free PMC article.
-
Macrophagic AMPKα1 orchestrates regenerative inflammation induced by glucocorticoids.EMBO Rep. 2023 Feb 6;24(2):e55363. doi: 10.15252/embr.202255363. Epub 2022 Dec 15. EMBO Rep. 2023. PMID: 36520372 Free PMC article.
-
Polyphenols and their effects on diabetes management: A review.Med J Islam Repub Iran. 2017 Dec 26;31:134. doi: 10.14196/mjiri.31.134. eCollection 2017. Med J Islam Repub Iran. 2017. PMID: 29951434 Free PMC article. Review.
-
The influence of goutweed (Aegopodium podagraria L.) tincture and metformin on the carbohydrate and lipid metabolism in dexamethasone-treated rats.BMC Complement Altern Med. 2016 Jul 22;16:235. doi: 10.1186/s12906-016-1221-y. BMC Complement Altern Med. 2016. PMID: 27450405 Free PMC article.
-
The PPARα pathway in Vγ9Vδ2 T cell anergy.Cell Mol Biol Lett. 2014 Dec;19(4):649-58. doi: 10.2478/s11658-014-0218-0. Epub 2014 Nov 25. Cell Mol Biol Lett. 2014. PMID: 25424910 Free PMC article.
References
-
- Barger PM, Browning AC, Garner AN, Kelly DP. p38 mitogen-activated protein kinase activates peroxisome proliferator-activated receptor alpha: a potential role in the cardiac metabolic stress response. J Biol Chem 276: 44495–44501, 2001 - PubMed
-
- Carey AL, Steinberg GR, Macaulay SL, Thomas WG, Holmes AG, Ramm G, Prelovsek O, Hohnen-Behrens C, Watt MJ, James DE, Kemp BE, Pedersen BK, Febbraio MA. Interleukin-6 increases insulin-stimulated glucose disposal in humans and glucose uptake and fatty acid oxidation in vitro via AMP-activated protein kinase. Diabetes 55: 2688–2697, 2006 - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical