

Increased gene sampling strengthens support for higher-level groups within leaf-mining moths and relatives (Lepidoptera: Gracillariidae)
- PMID: 21702958
- PMCID: PMC3145599
- DOI: 10.1186/1471-2148-11-182
Increased gene sampling strengthens support for higher-level groups within leaf-mining moths and relatives (Lepidoptera: Gracillariidae)
Abstract
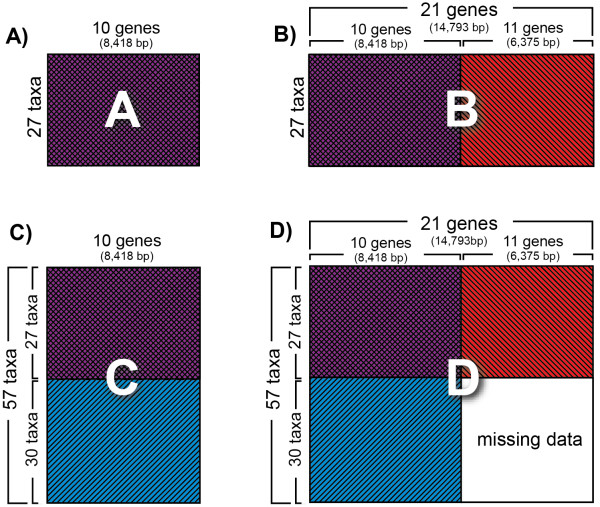
Background: Researchers conducting molecular phylogenetic studies are frequently faced with the decision of what to do when weak branch support is obtained for key nodes of importance. As one solution, the researcher may choose to sequence additional orthologous genes of appropriate evolutionary rate for the taxa in the study. However, generating large, complete data matrices can become increasingly difficult as the number of characters increases. A few empirical studies have shown that augmenting genes even for a subset of taxa can improve branch support. However, because each study differs in the number of characters and taxa, there is still a need for additional studies that examine whether incomplete sampling designs are likely to aid at increasing deep node resolution. We target Gracillariidae, a Cretaceous-age (~100 Ma) group of leaf-mining moths to test whether the strategy of adding genes for a subset of taxa can improve branch support for deep nodes. We initially sequenced ten genes (8,418 bp) for 57 taxa that represent the major lineages of Gracillariidae plus outgroups. After finding that many deep divergences remained weakly supported, we sequenced eleven additional genes (6,375 bp) for a 27-taxon subset. We then compared results from different data sets to assess whether one sampling design can be favored over another. The concatenated data set comprising all genes and all taxa and three other data sets of different taxon and gene sub-sampling design were analyzed with maximum likelihood. Each data set was subject to five different models and partitioning schemes of non-synonymous and synonymous changes. Statistical significance of non-monophyly was examined with the Approximately Unbiased (AU) test.
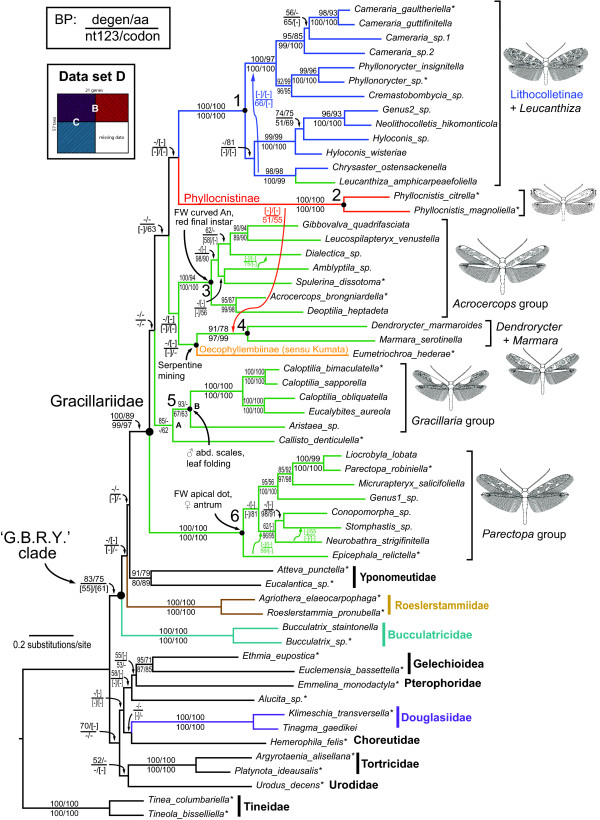
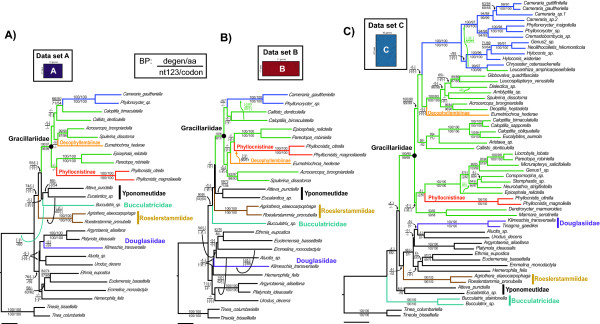
Results: Partial augmentation of genes led to high support for deep divergences, especially when non-synonymous changes were analyzed alone. Increasing the number of taxa without an increase in number of characters led to lower bootstrap support; increasing the number of characters without increasing the number of taxa generally increased bootstrap support. More than three-quarters of nodes were supported with bootstrap values greater than 80% when all taxa and genes were combined. Gracillariidae, Lithocolletinae + Leucanthiza, and Acrocercops and Parectopa groups were strongly supported in nearly every analysis. Gracillaria group was well supported in some analyses, but less so in others. We find strong evidence for the exclusion of Douglasiidae from Gracillarioidea sensu Davis and Robinson (1998). Our results strongly support the monophyly of a G.B.R.Y. clade, a group comprised of Gracillariidae + Bucculatricidae + Roeslerstammiidae + Yponomeutidae, when analyzed with non-synonymous changes only, but this group was frequently split when synonymous and non-synonymous substitutions were analyzed together.
Conclusions: 1) Partially or fully augmenting a data set with more characters increased bootstrap support for particular deep nodes, and this increase was dramatic when non-synonymous changes were analyzed alone. Thus, the addition of sites that have low levels of saturation and compositional heterogeneity can greatly improve results. 2) Gracillarioidea, as defined by Davis and Robinson (1998), clearly do not include Douglasiidae, and changes to current classification will be required. 3) Gracillariidae were monophyletic in all analyses conducted, and nearly all species can be placed into one of six strongly supported clades though relationships among these remain unclear. 4) The difficulty in determining the phylogenetic placement of Bucculatricidae is probably attributable to compositional heterogeneity at the third codon position. From our tests for compositional heterogeneity and strong bootstrap values obtained when synonymous changes are excluded, we tentatively conclude that Bucculatricidae is closely related to Gracillariidae + Roeslerstammiidae + Yponomeutidae.
Figures
Similar articles
-
A large-scale, higher-level, molecular phylogenetic study of the insect order Lepidoptera (moths and butterflies).PLoS One. 2013;8(3):e58568. doi: 10.1371/journal.pone.0058568. Epub 2013 Mar 12. PLoS One. 2013. PMID: 23554903 Free PMC article.
-
Can deliberately incomplete gene sample augmentation improve a phylogeny estimate for the advanced moths and butterflies (Hexapoda: Lepidoptera)?Syst Biol. 2011 Dec;60(6):782-96. doi: 10.1093/sysbio/syr079. Epub 2011 Aug 16. Syst Biol. 2011. PMID: 21840842 Free PMC article.
-
Diversity of Australian Ornixolinae (Lepidoptera: Gracillariidae) with taxonomic and nomenclatural acts within the related taxa (Acrocercopinae and Gracillariinae) based on the evidence of museomics, bionomics, and mitogenomics.Zootaxa. 2025 Mar 31;5616(1):1-340. doi: 10.11646/zootaxa.5616.1.1. Zootaxa. 2025. PMID: 40173480
-
The impact of taxon sampling on phylogenetic inference: a review of two decades of controversy.Brief Bioinform. 2012 Jan;13(1):122-34. doi: 10.1093/bib/bbr014. Epub 2011 Mar 23. Brief Bioinform. 2012. PMID: 21436145 Free PMC article. Review.
-
Can We Reliably Calibrate Deep Nodes in the Tetrapod Tree? Case Studies in Deep Tetrapod Divergences.Front Genet. 2020 Oct 16;11:506749. doi: 10.3389/fgene.2020.506749. eCollection 2020. Front Genet. 2020. PMID: 33193596 Free PMC article. Review.
Cited by
-
A molecular phylogeny for the leaf-roller moths (Lepidoptera: Tortricidae) and its implications for classification and life history evolution.PLoS One. 2012;7(4):e35574. doi: 10.1371/journal.pone.0035574. Epub 2012 Apr 19. PLoS One. 2012. PMID: 22536410 Free PMC article.
-
Novel insight into lepidopteran phylogenetics from the mitochondrial genome of the apple fruit moth of the family Argyresthiidae.BMC Genomics. 2024 Jan 2;25(1):21. doi: 10.1186/s12864-023-09905-1. BMC Genomics. 2024. PMID: 38166583 Free PMC article.
-
A molecular phylogeny for yponomeutoidea (insecta, Lepidoptera, ditrysia) and its implications for classification, biogeography and the evolution of host plant use.PLoS One. 2013;8(1):e55066. doi: 10.1371/journal.pone.0055066. Epub 2013 Jan 31. PLoS One. 2013. PMID: 23383061 Free PMC article.
-
DNA Barcodes Combined with Multilocus Data of Representative Taxa Can Generate Reliable Higher-Level Phylogenies.Syst Biol. 2022 Feb 10;71(2):382-395. doi: 10.1093/sysbio/syab038. Syst Biol. 2022. PMID: 34022059 Free PMC article.
-
A large-scale, higher-level, molecular phylogenetic study of the insect order Lepidoptera (moths and butterflies).PLoS One. 2013;8(3):e58568. doi: 10.1371/journal.pone.0058568. Epub 2013 Mar 12. PLoS One. 2013. PMID: 23554903 Free PMC article.
References
-
- Bapteste E, Brinkmann H, Lee JA, Moore DV, Sensen CW, Gordon P, Duruflé L, Gaasterland T, Lopez P, Müller M. et al.The analysis of 100 genes supports the grouping of three highly divergent amoebae: Dictyostelium, Entamoeba, and Mastigamoeba. Proceedings of the National Academy of Sciences, USA. 2002;99:1414–1419. doi: 10.1073/pnas.032662799. - DOI - PMC - PubMed
-
- Zwick A, Regier JC, Mitter C, Cummings MP. Increased gene sampling yields robust support for higher-level clades within Bombycoidea (Lepidoptera) Systematic Entomology. 2011;36:31–43. doi: 10.1111/j.1365-3113.2010.00543.x. - DOI
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
