

Restoring dystrophin expression in duchenne muscular dystrophy muscle progress in exon skipping and stop codon read through
- PMID: 21703390
- PMCID: PMC3124804
- DOI: 10.1016/j.ajpath.2011.03.050
Restoring dystrophin expression in duchenne muscular dystrophy muscle progress in exon skipping and stop codon read through
Abstract
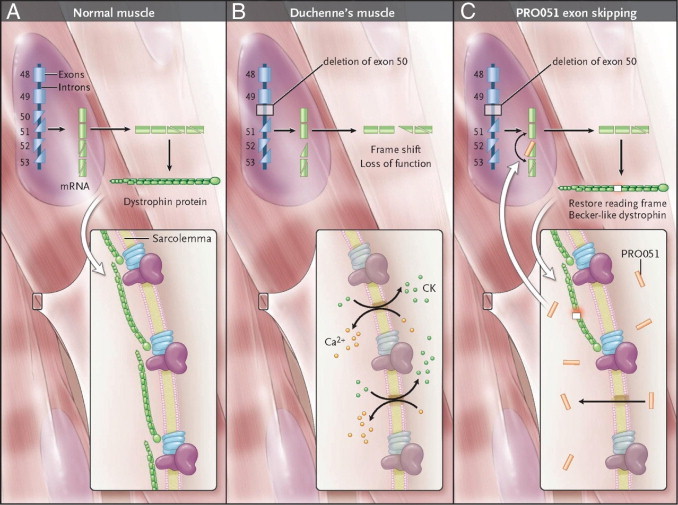
The identification of the Duchenne muscular dystrophy gene and protein in the late 1980s led to high hopes of rapid translation to molecular therapeutics. These hopes were fueled by early reports of delivering new functional genes to dystrophic muscle in mouse models using gene therapy and stem cell transplantation. However, significant barriers have thwarted translation of these approaches to true therapies, including insufficient therapeutic material (eg, cells and viral vectors), challenges in systemic delivery, and immunological hurdles. An alternative approach is to repair the patient's own gene. Two innovative small-molecule approaches have emerged as front-line molecular therapeutics: exon skipping and stop codon read through. Both approaches are in human clinical trials and aim to coax dystrophin protein production from otherwise inactive mutant genes. In the clinically severe dog model of Duchenne muscular dystrophy, the exon-skipping approach recently improved multiple functional outcomes. We discuss the status of these two methods aimed at inducing de novo dystrophin production from mutant genes and review implications for other disorders.
Copyright © 2011 American Society for Investigative Pathology. Published by Elsevier Inc. All rights reserved.
Figures



References
-
- Hoffman E.P., Brown R.H., Jr, Kunkel L.M. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51:919–928. - PubMed
-
- Hoffman E.P., Hudecki M.S., Rosenberg P.A., Pollina C.M., Kunkel L.M. Cell and fiber-type distribution of dystrophin. Neuron. 1988;1:411–420. - PubMed
-
- Koenig M., Hoffman E.P., Bertelson C.J., Monaco A.P., Feener C., Kunkel L.M. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell. 1987;50:509–517. - PubMed
-
- Hoffman E.P., Fischbeck K.H., Brown R.H., Johnson M., Medori R., Loike J.D., Harris J.B., Waterston R., Brooke M., Specht L., Kupsky W., Chamberlain J., Caskey C.T., Shapiro F., Kunkel L.M. Characterization of dystrophin in muscle-biopsy specimens from patients with Duchenne's or Becker's muscular dystrophy. N Engl J Med. 1988;318:1363–1368. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
