

Studies on human DNA polymerase epsilon and GINS complex and their role in DNA replication
- PMID: 21705323
- PMCID: PMC3190704
- DOI: 10.1074/jbc.M111.256289
Studies on human DNA polymerase epsilon and GINS complex and their role in DNA replication
Abstract
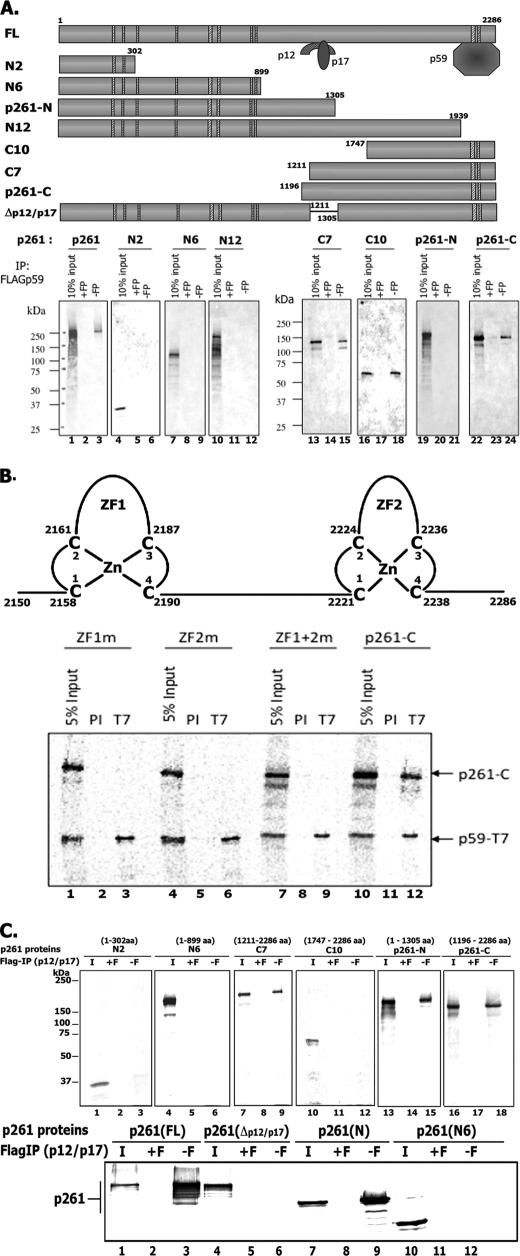
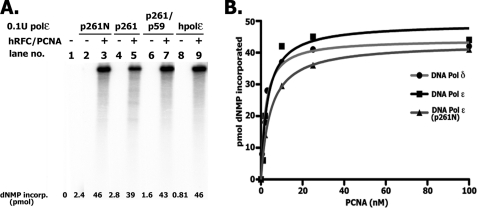
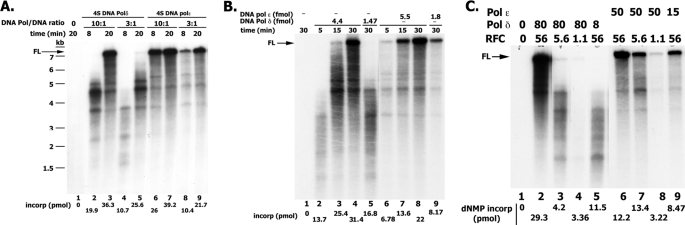
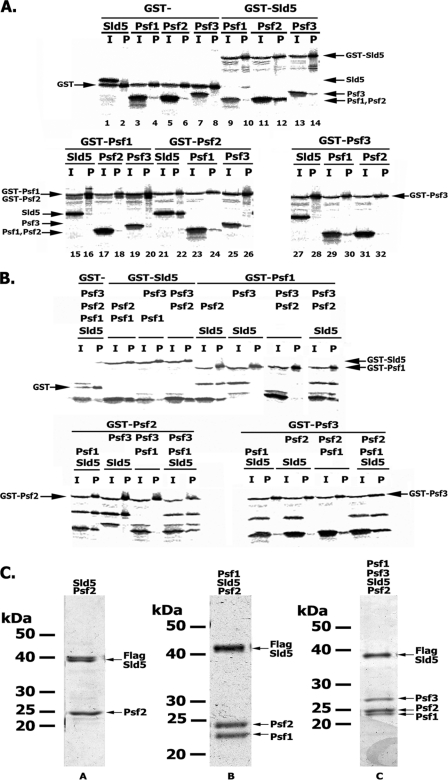
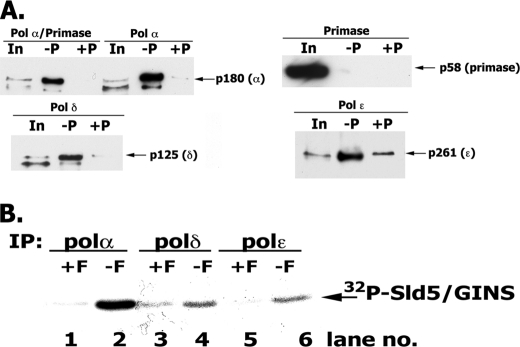
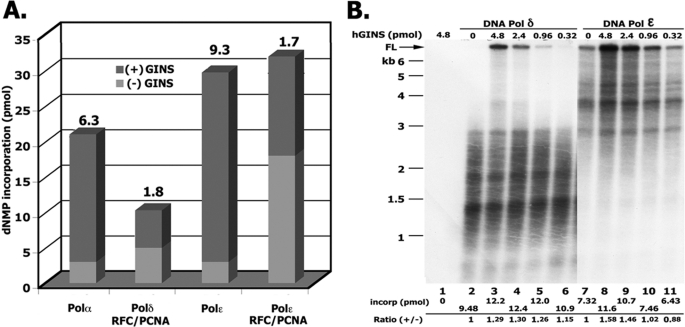
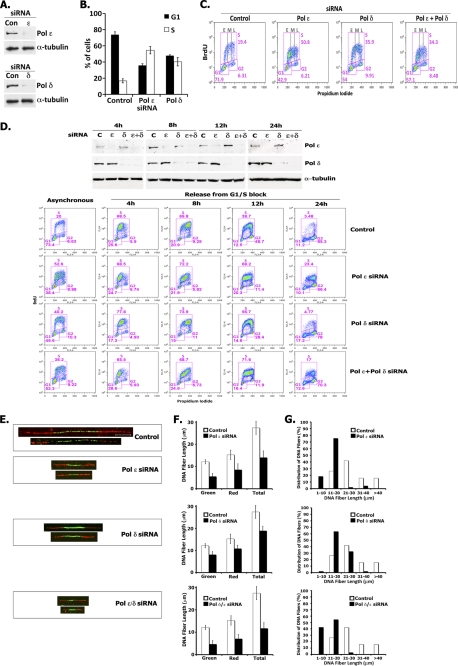
In eukaryotic cells, DNA replication is carried out by the coordinated action of three DNA polymerases (Pols), Pol α, δ, and ε. In this report, we describe the reconstitution of the human four-subunit Pol ε and characterization of its catalytic properties in comparison with Pol α and Pol δ. Human Pol ε holoenzyme is a monomeric complex containing stoichiometric subunit levels of p261/Pol 2, p59, p17, and p12. We show that the Pol ε p261 N-terminal catalytic domain is solely responsible for its ability to catalyze DNA synthesis. Importantly, human Pol (hPol) ε was found more processive than hPol δ in supporting proliferating cell nuclear antigen-dependent elongation of DNA chains, which is in keeping with proposed roles for hPol ε and hPol δ in the replication of leading and lagging strands, respectively. Furthermore, GINS, a component of the replicative helicase complex that is composed of Sld5, Psf1, Psf2, and Psf3, was shown to interact weakly with all three replicative DNA Pols (α, δ, and ε) and to markedly stimulate the activities of Pol α and Pol ε. In vivo studies indicated that siRNA-targeted depletion of hPol δ and/or hPol ε reduced cell cycle progression and the rate of fork progression. Under the conditions used, we noted that depletion of Pol ε had a more pronounced inhibitory effect on cellular DNA replication than depletion of Pol δ. We suggest that reduction in the level of Pol δ may be less deleterious because of its collision-and-release role in lagging strand synthesis.
Figures

References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
