

Increased methylation variation in epigenetic domains across cancer types
- PMID: 21706001
- PMCID: PMC3145050
- DOI: 10.1038/ng.865
Increased methylation variation in epigenetic domains across cancer types
Abstract
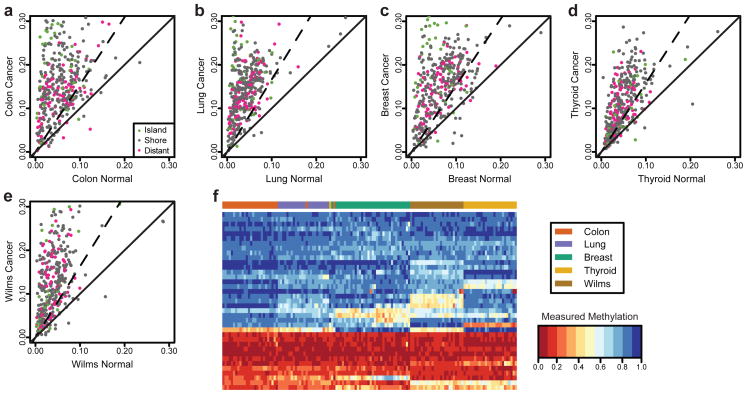
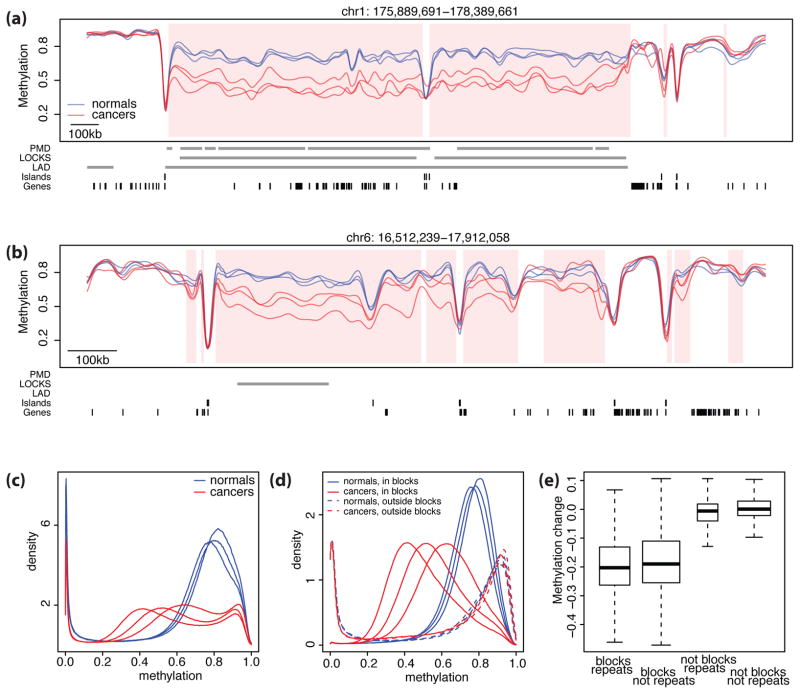
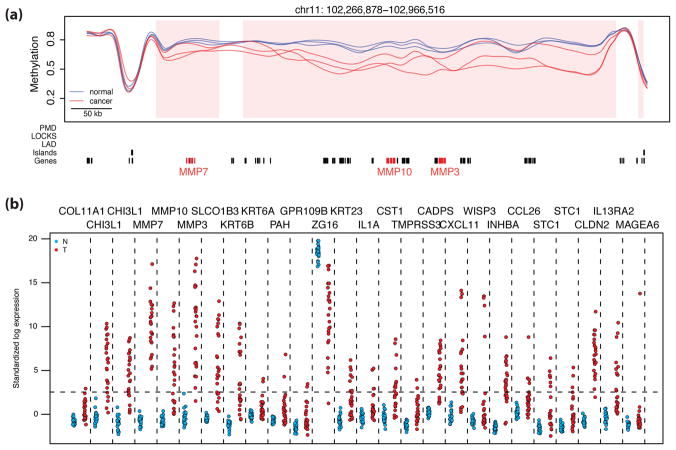
Tumor heterogeneity is a major barrier to effective cancer diagnosis and treatment. We recently identified cancer-specific differentially DNA-methylated regions (cDMRs) in colon cancer, which also distinguish normal tissue types from each other, suggesting that these cDMRs might be generalized across cancer types. Here we show stochastic methylation variation of the same cDMRs, distinguishing cancer from normal tissue, in colon, lung, breast, thyroid and Wilms' tumors, with intermediate variation in adenomas. Whole-genome bisulfite sequencing shows these variable cDMRs are related to loss of sharply delimited methylation boundaries at CpG islands. Furthermore, we find hypomethylation of discrete blocks encompassing half the genome, with extreme gene expression variability. Genes associated with the cDMRs and large blocks are involved in mitosis and matrix remodeling, respectively. We suggest a model for cancer involving loss of epigenetic stability of well-defined genomic domains that underlies increased methylation variability in cancer that may contribute to tumor heterogeneity.
Figures


Comment in
-
Epigenetic variation and cellular Darwinism.Nat Genet. 2011 Jul 27;43(8):724-6. doi: 10.1038/ng.897. Nat Genet. 2011. PMID: 21792236 No abstract available.
References
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
Grants and funding
- R37 CA054358/CA/NCI NIH HHS/United States
- P50 HG003233/HG/NHGRI NIH HHS/United States
- F32CA138111/CA/NCI NIH HHS/United States
- R01 HG006102/HG/NHGRI NIH HHS/United States
- R01 GM083084/GM/NIGMS NIH HHS/United States
- R01HG005220/HG/NHGRI NIH HHS/United States
- R01DA025779/DA/NIDA NIH HHS/United States
- R01 HG005220/HG/NHGRI NIH HHS/United States
- F32 CA138111/CA/NCI NIH HHS/United States
- 5R01GM083084/GM/NIGMS NIH HHS/United States
- 5P50HG003233/HG/NHGRI NIH HHS/United States
- R01 DA025779/DA/NIDA NIH HHS/United States
- R01 CA054358/CA/NCI NIH HHS/United States
- R37CA054358/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
