

Direct measurement of DNA affinity landscapes on a high-throughput sequencing instrument
- PMID: 21706015
- PMCID: PMC3134637
- DOI: 10.1038/nbt.1882
Direct measurement of DNA affinity landscapes on a high-throughput sequencing instrument
Abstract
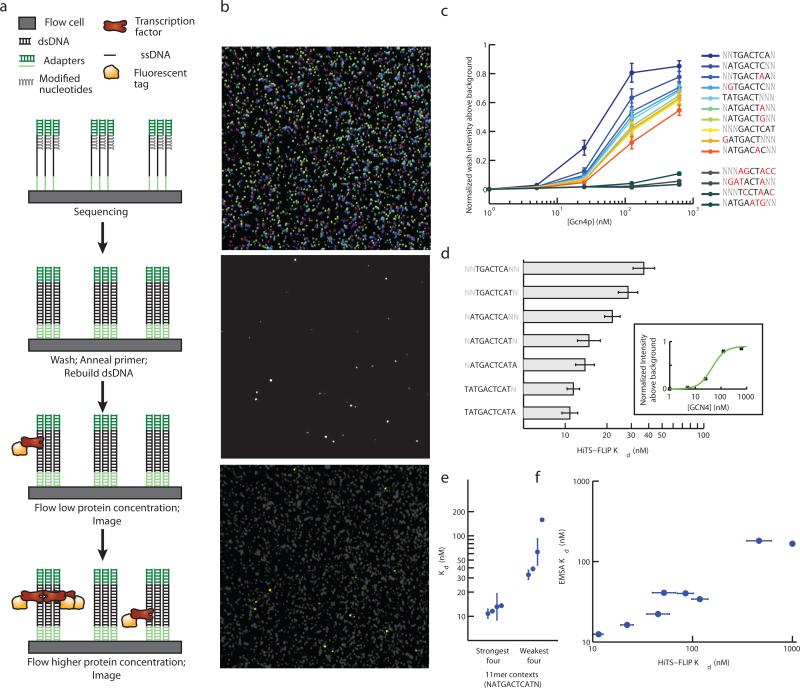
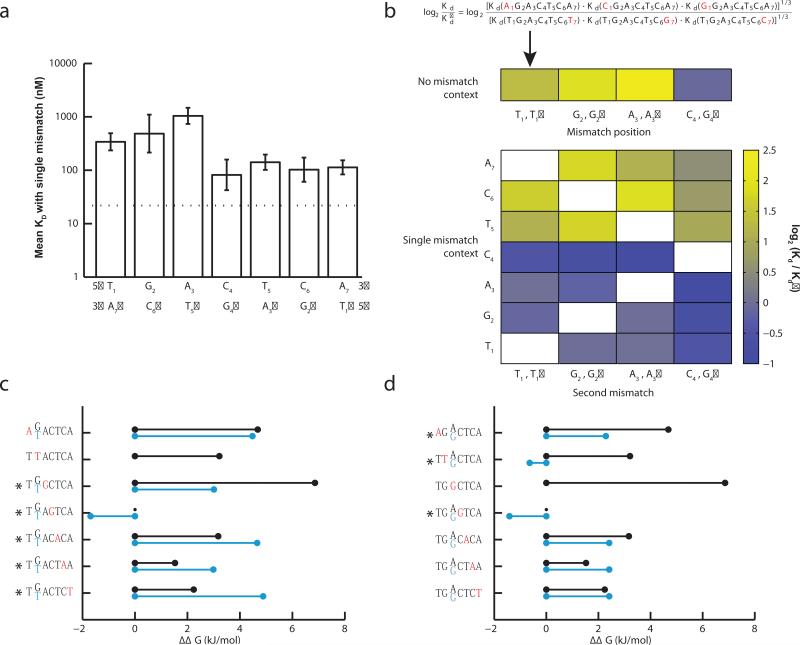
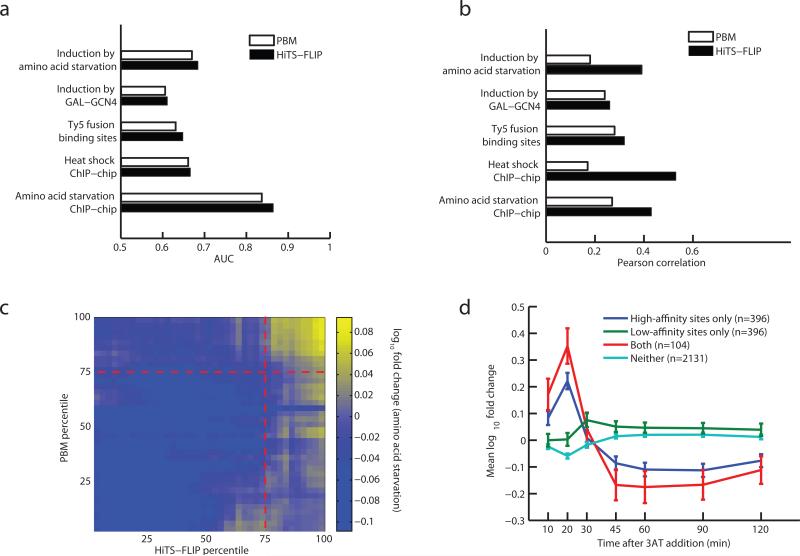
Several methods for characterizing DNA-protein interactions are available, but none have demonstrated both high throughput and quantitative measurement of affinity. Here we describe 'high-throughput sequencing'-'fluorescent ligand interaction profiling' (HiTS-FLIP), a technique for measuring quantitative protein-DNA binding affinity at unprecedented depth. In this approach, the optics built into a high-throughput sequencer are used to visualize in vitro binding of a protein to sequenced DNA in a flow cell. Application of HiTS-FLIP to the protein Gcn4 (Gcn4p), the master regulator of the yeast amino acid starvation response, yielded ~440 million binding measurements, enabling determination of dissociation constants for all 12-mer sequences having submicromolar affinity. These data revealed a complex interdependency between motif positions, allowed improved discrimination of in vivo Gcn4p binding sites and regulatory targets relative to previous methods and showed that sets of genes with different promoter affinities to Gcn4p have distinct functions and expression kinetics. Broad application of this approach should increase understanding of the interactions that drive transcription.
Figures
References
-
- Klug SJ, Famulok M. All you wanted to know about SELEX. Mol Biol Rep. 1994;20:97–107. - PubMed
-
- Ren B, et al. Genome-wide location and function of DNA binding proteins. Science. 2000;290:2306–2309. - PubMed
-
- Johnson DS, Mortazavi A, Myers RM, Wold B. Genome-wide mapping of in vivo protein-DNA interactions. Science. 2007;316:1497–1502. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
