

Modes of Aβ toxicity in Alzheimer's disease
- PMID: 21706148
- PMCID: PMC3181413
- DOI: 10.1007/s00018-011-0750-2
Modes of Aβ toxicity in Alzheimer's disease
Abstract
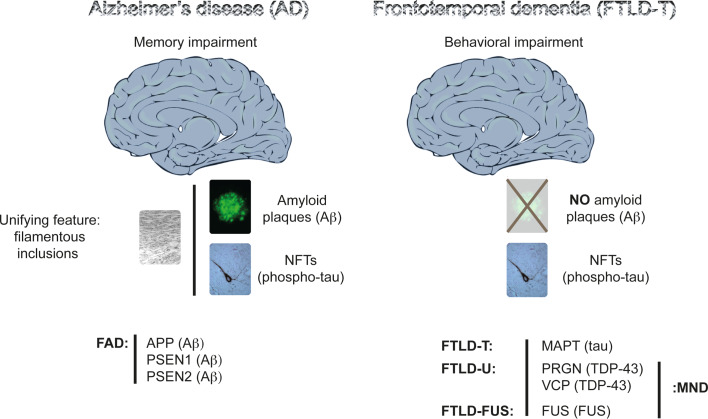
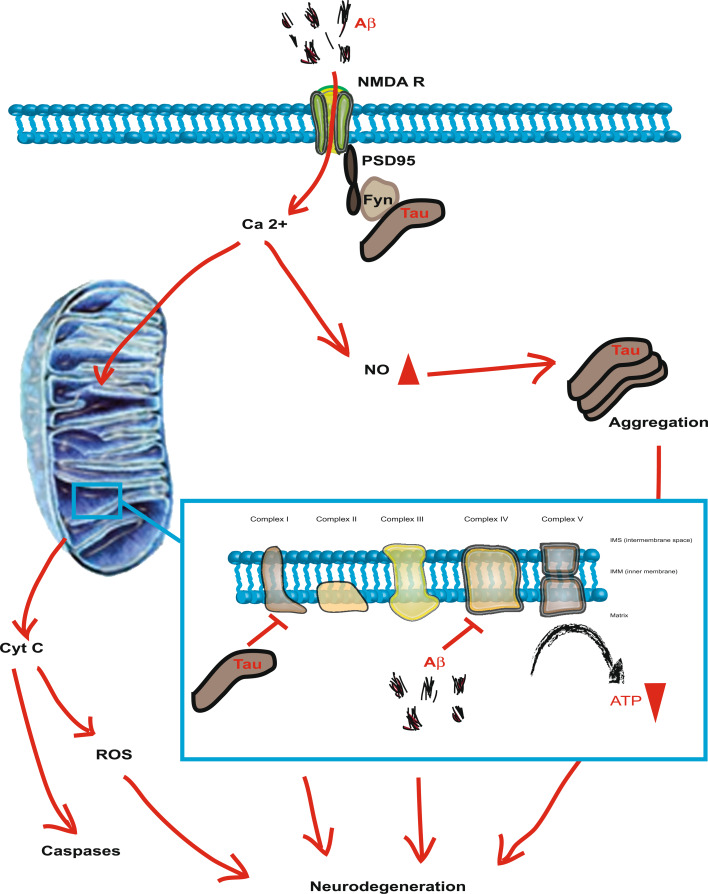
Alzheimer's disease (AD) is reaching epidemic proportions, yet a cure is not yet available. While the genetic causes of the rare familial inherited forms of AD are understood, the causes of the sporadic forms of the disease are not. Histopathologically, these two forms of AD are indistinguishable: they are characterized by amyloid-β (Aβ) peptide-containing amyloid plaques and tau-containing neurofibrillary tangles. In this review we compare AD to frontotemporal dementia (FTD), a subset of which is characterized by tau deposition in the absence of overt plaques. A host of transgenic animal AD models have been established through the expression of human proteins with pathogenic mutations previously identified in familial AD and FTD. Determining how these mutant proteins cause disease in vivo should contribute to an understanding of the causes of the more frequent sporadic forms. We discuss the insight transgenic animal models have provided into Aβ and tau toxicity, also with regards to mitochondrial function and the crucial role tau plays in mediating Aβ toxicity. We also discuss the role of miRNAs in mediating the toxic effects of the Aβ peptide.
Figures
Similar articles
-
Amyloid-induced neurofibrillary tangle formation in Alzheimer's disease: insight from transgenic mouse and tissue-culture models.Int J Dev Neurosci. 2004 Nov;22(7):453-65. doi: 10.1016/j.ijdevneu.2004.07.013. Int J Dev Neurosci. 2004. PMID: 15465275 Review.
-
Alzheimer's disease and frontotemporal dementia: prospects of a tailored therapy?Med J Aust. 2006 Oct 2;185(7):381-4. doi: 10.5694/j.1326-5377.2006.tb00615.x. Med J Aust. 2006. PMID: 17014407 Review.
-
Aβ-induced acceleration of Alzheimer-related τ-pathology spreading and its association with prion protein.Acta Neuropathol. 2019 Dec;138(6):913-941. doi: 10.1007/s00401-019-02053-5. Epub 2019 Aug 14. Acta Neuropathol. 2019. PMID: 31414210
-
Molecular and functional signatures in a novel Alzheimer's disease mouse model assessed by quantitative proteomics.Mol Neurodegener. 2018 Jan 16;13(1):2. doi: 10.1186/s13024-017-0234-4. Mol Neurodegener. 2018. PMID: 29338754 Free PMC article.
-
Active full-length DNA Aβ42 immunization in 3xTg-AD mice reduces not only amyloid deposition but also tau pathology.Alzheimers Res Ther. 2018 Nov 20;10(1):115. doi: 10.1186/s13195-018-0441-4. Alzheimers Res Ther. 2018. PMID: 30454039 Free PMC article.
Cited by
-
Dysregulation of glutathione homeostasis in neurodegenerative diseases.Nutrients. 2012 Oct 9;4(10):1399-440. doi: 10.3390/nu4101399. Nutrients. 2012. PMID: 23201762 Free PMC article. Review.
-
Curcumin and Apigenin - novel and promising therapeutics against chronic neuroinflammation in Alzheimer's disease.Neural Regen Res. 2015 Aug;10(8):1181-5. doi: 10.4103/1673-5374.162686. Neural Regen Res. 2015. PMID: 26487830 Free PMC article. Review.
-
Inhibition of the mitochondrial enzyme ABAD restores the amyloid-β-mediated deregulation of estradiol.PLoS One. 2011;6(12):e28887. doi: 10.1371/journal.pone.0028887. Epub 2011 Dec 12. PLoS One. 2011. PMID: 22174920 Free PMC article.
-
Unfolded protein response-induced ERdj3 secretion links ER stress to extracellular proteostasis.EMBO J. 2015 Jan 2;34(1):4-19. doi: 10.15252/embj.201488896. Epub 2014 Oct 31. EMBO J. 2015. PMID: 25361606 Free PMC article.
-
Tau45-230 association with the cytoskeleton and membrane-bound organelles: Functional implications in neurodegeneration.Neuroscience. 2017 Oct 24;362:104-117. doi: 10.1016/j.neuroscience.2017.08.026. Epub 2017 Aug 24. Neuroscience. 2017. PMID: 28844006 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
