

Predicting a small molecule-kinase interaction map: A machine learning approach
- PMID: 21708012
- PMCID: PMC3151211
- DOI: 10.1186/1758-2946-3-22
Predicting a small molecule-kinase interaction map: A machine learning approach
Abstract
Background: We present a machine learning approach to the problem of protein ligand interaction prediction. We focus on a set of binding data obtained from 113 different protein kinases and 20 inhibitors. It was attained through ATP site-dependent binding competition assays and constitutes the first available dataset of this kind. We extract information about the investigated molecules from various data sources to obtain an informative set of features.
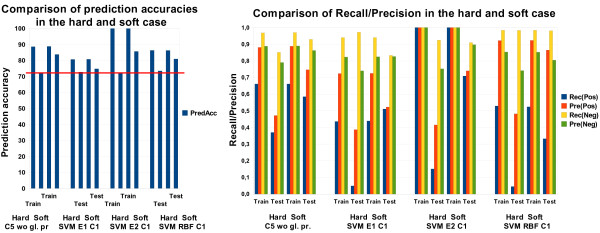
Results: A Support Vector Machine (SVM) as well as a decision tree algorithm (C5/See5) is used to learn models based on the available features which in turn can be used for the classification of new kinase-inhibitor pair test instances. We evaluate our approach using different feature sets and parameter settings for the employed classifiers. Moreover, the paper introduces a new way of evaluating predictions in such a setting, where different amounts of information about the binding partners can be assumed to be available for training. Results on an external test set are also provided.
Conclusions: In most of the cases, the presented approach clearly outperforms the baseline methods used for comparison. Experimental results indicate that the applied machine learning methods are able to detect a signal in the data and predict binding affinity to some extent. For SVMs, the binding prediction can be improved significantly by using features that describe the active site of a kinase. For C5, besides diversity in the feature set, alignment scores of conserved regions turned out to be very useful.
Figures









References
-
- LaValle SM, Finn PW, Kavraki LE, Latombe JC. Efficient database screening for rational drug design using pharmacophore-constrained conformational search. Proceedings of the third annual international conference on computational molecular biology, RECOMB'99, April 11-14, Lyon, France. 1999. pp. 250–260.
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
