

Molecular diagnosis of hereditary inclusion body myopathy by linkage analysis and identification of a novel splice site mutation in GNE
- PMID: 21708040
- PMCID: PMC3141630
- DOI: 10.1186/1471-2350-12-87
Molecular diagnosis of hereditary inclusion body myopathy by linkage analysis and identification of a novel splice site mutation in GNE
Abstract
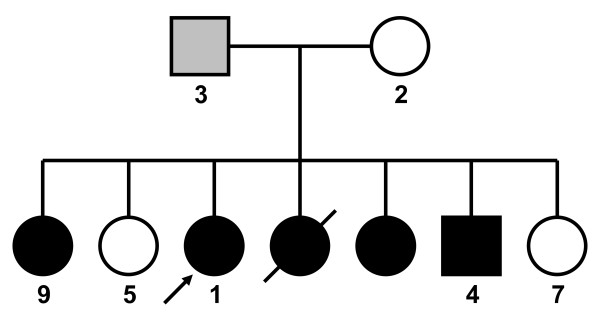
Background: Many myopathies share clinical features in common, and diagnosis often requires genetic testing. We ascertained a family in which five siblings presented with distal muscle weakness of unknown etiology.
Methods: We performed high-density genomewide linkage analysis and mutation screening of candidate genes to identify the genetic defect in the family. Preserved clinical biopsy material was reviewed to confirm the diagnosis, and reverse transcriptase PCR was used to determine the molecular effect of a splice site mutation.
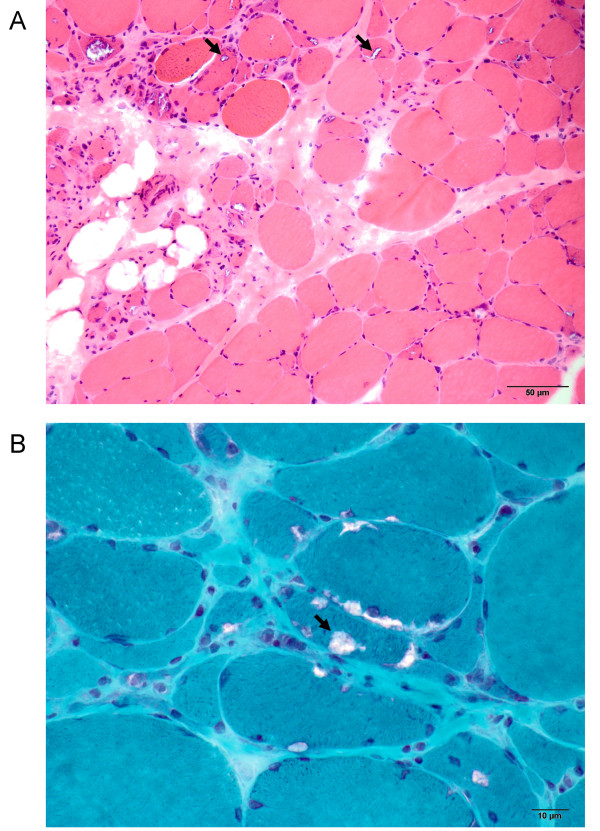
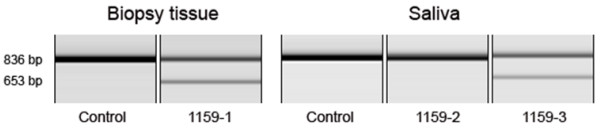
Results: The linkage scan excluded the majority of known myopathy genes, but one linkage peak included the gene GNE, in which mutations cause autosomal recessive hereditary inclusion body myopathy type 2 (HIBM2). Muscle biopsy tissue from a patient showed myopathic features, including small basophilic fibers with vacuoles. Sequence analysis of GNE revealed affected individuals were compound heterozygous for a novel mutation in the 5' splice donor site of intron 10 (c.1816+5G>A) and a previously reported missense mutation (c.2086G>A, p.V696M), confirming the diagnosis as HIBM2. The splice site mutation correlated with exclusion of exon 10 from the transcript, which is predicted to produce an in-frame deletion (p.G545_D605del) of 61 amino acids in the kinase domain of the GNE protein. The father of the proband was heterozygous for the splice site mutation and exhibited mild distal weakness late in life.
Conclusions: Our study expands on the extensive allelic heterogeneity of HIBM2 and demonstrates the value of linkage analysis in resolving ambiguous clinical findings to achieve a molecular diagnosis.
Figures
References
-
- Neudecker S, Krasnianski M, Bahn E, Zierz S. Rimmed vacuoles in facioscapulohumeral muscular dystrophy: a unique ultrastructural feature. Acta Neuropathol. 2004;108(3):257–259. - PubMed
-
- Hauser MA, Horrigan SK, Salmikangas P, Torian UM, Viles KD, Dancel R, Tim RW, Taivainen A, Bartoloni L, Gilchrist JM, Stajich JM, Gaskell PC, Gilbert JR, Vance JM, Pericak-Vance MA, Carpen O, Westbrook CA, Speer MC. Myotilin is mutated in limb girdle muscular dystrophy 1A. Hum Mol Genet. 2000;9(14):2141–2147. doi: 10.1093/hmg/9.14.2141. - DOI - PubMed
-
- Eisenberg I, Avidan N, Potikha T, Hochner H, Chen M, Olender T, Barash M, Shemesh M, Sadeh M, Grabov-Nardini G, Shmilevich I, Friedmann A, Karpati G, Bradley WG, Baumbach L, Lancet D, Asher EB, Beckmann JS, Argov Z, Mitrani-Rosenbaum S. The UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase gene is mutated in recessive hereditary inclusion body myopathy. Nat Genet. 2001;29(1):83–87. doi: 10.1038/ng718. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
