

Orphan missense mutations in the cystic fibrosis transmembrane conductance regulator: A three-step biological approach to establishing a correlation between genotype and phenotype
- PMID: 21708286
- PMCID: PMC3157621
- DOI: 10.1016/j.jmoldx.2011.05.004
Orphan missense mutations in the cystic fibrosis transmembrane conductance regulator: A three-step biological approach to establishing a correlation between genotype and phenotype
Abstract
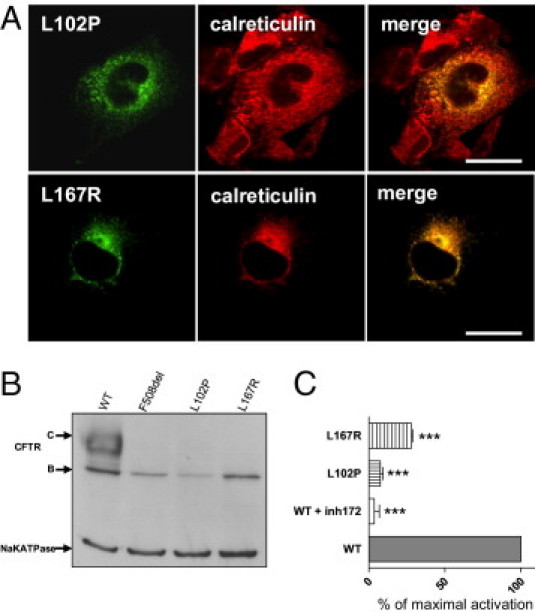
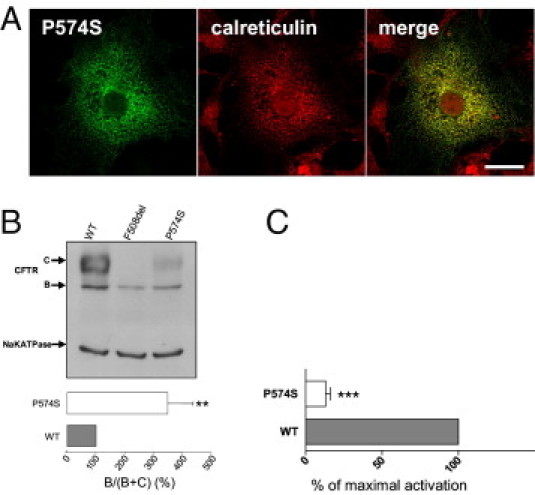
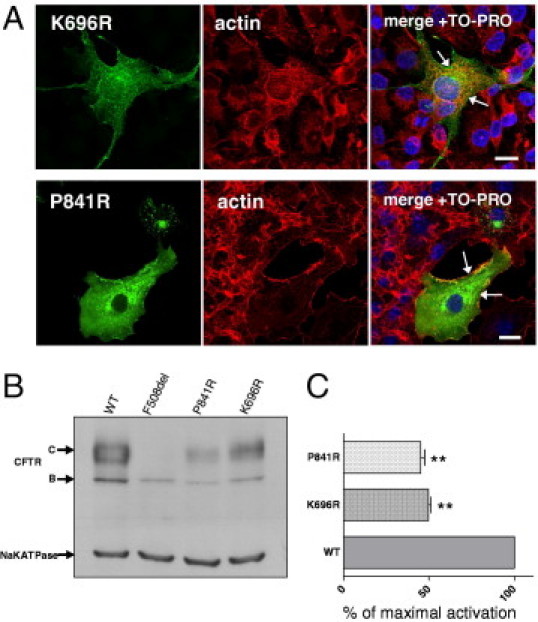
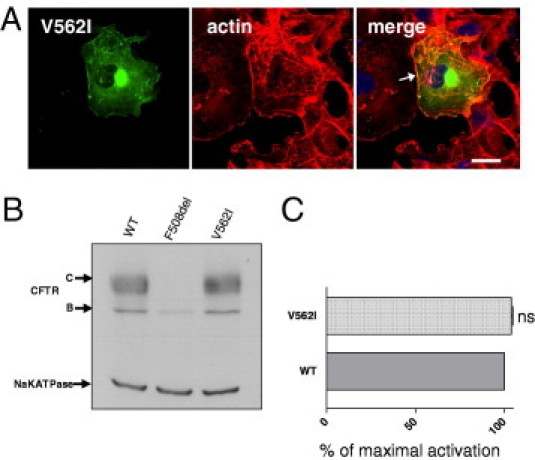
More than 1860 mutations have been found within the human cystic fibrosis transmembrane conductance regulator (CFTR) gene sequence. These mutations can be classified according to their degree of severity in CF disease. Although the most common mutations are well characterized, few data are available for rare mutations. Thus, genetic counseling is particularly difficult when fetuses or patients with CF present these orphan variations. We describe a three-step in vitro assay that can evaluate rare missense CFTR mutation consequences to establish a correlation between genotype and phenotype. By using a green fluorescent protein-tagged CFTR construct, we expressed mutated proteins in COS-7 cells. CFTR trafficking was visualized by confocal microscopy, and the cellular localization of CFTR was determined using intracellular markers. We studied the CFTR maturation process using Western blot analysis and evaluated CFTR channel activity by automated iodide efflux assays. Of six rare mutations that we studied, five have been isolated in our laboratory. The cellular and functional impact that we observed in each case was compared with the clinical data concerning the patients in whom we encountered these mutations. In conclusion, we propose that performing this type of analysis for orphan CFTR missense mutations can improve CF genetic counseling.
Copyright © 2011 American Society for Investigative Pathology and the Association for Molecular Pathology. Published by Elsevier Inc. All rights reserved.
Figures

Similar articles
-
Molecular and functional characterization of CBAVD-causing mutations located in CFTR nucleotide-binding domains.Cell Physiol Biochem. 2008;22(1-4):79-92. doi: 10.1159/000149785. Epub 2008 Jul 25. Cell Physiol Biochem. 2008. PMID: 18769034
-
Localization studies of rare missense mutations in cystic fibrosis transmembrane conductance regulator (CFTR) facilitate interpretation of genotype-phenotype relationships.Hum Mutat. 2008 Nov;29(11):1364-72. doi: 10.1002/humu.20866. Hum Mutat. 2008. PMID: 18951463 Free PMC article.
-
Effects of cystic fibrosis and congenital bilateral absence of the vas deferens-associated mutations on cystic fibrosis transmembrane conductance regulator-mediated regulation of separate channels.Am J Hum Genet. 2000 May;66(5):1485-95. doi: 10.1086/302893. Epub 2000 Apr 4. Am J Hum Genet. 2000. PMID: 10762539 Free PMC article.
-
Genotype and phenotype in cystic fibrosis.Respiration. 2000;67(2):117-33. doi: 10.1159/000029497. Respiration. 2000. PMID: 10773783 Review.
-
Molecular Genetics of Cystic Fibrosis Transmembrane Conductance Regulator: Genotype and Phenotype.Pediatr Clin North Am. 2016 Aug;63(4):585-98. doi: 10.1016/j.pcl.2016.04.002. Pediatr Clin North Am. 2016. PMID: 27469177 Review.
Cited by
-
CFTR-Related Metabolic Syndrome: Genetic Variants Increasing Pancreatitis Risk in the Pediatric Puerto Rican Population.Children (Basel). 2023 Jan 31;10(2):280. doi: 10.3390/children10020280. Children (Basel). 2023. PMID: 36832409 Free PMC article.
References
-
- Riordan J.R., Rommens J.M., Kerem B.S., Alon N., Rozmahel R., Grzelczak Z., Zielenski J., Lok S., Plavsic N., Chou J.L., Drumm M.L., Iannuzzi M.C., Collins F.S., Tsui L.C. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245:1066–1073. - PubMed
-
- Rommens J.M., Iannuzzi C., Kerem B.S., Drumm M.L., Melmer G., Dean M., Rozmahel R., Cole J.L., Kennedy D., Hidaka N., Zsiga M., Buchwald M., Riordan J.R., Tsui L.C., Collins F.S. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science. 1989;245:1059–1065. - PubMed
-
- Riordan J.R. CFTR function and prospects for therapy. Annu Rev Biochem. 2008;77:701–726. - PubMed
-
- Lommatzsch S.T., Aris R. Genetics of cystic fibrosis. Semin Respir Crit Care Med. 2009;30:531–538. - PubMed
-
- Linton K.J., Rosenberg M.F., Kerr I.D., Higgins C.F. Structure of ABC transporters: ABC Proteins: From Bacteria to Man. In: Holland I.B., Cole S., Kuchler K., Higgins C.F., editors. Academic Press; Waltham, MA: 2003. pp. 65–80.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
