

Host shifts and molecular evolution of H7 avian influenza virus hemagglutinin
- PMID: 21711553
- PMCID: PMC3141685
- DOI: 10.1186/1743-422X-8-328
Host shifts and molecular evolution of H7 avian influenza virus hemagglutinin
Abstract
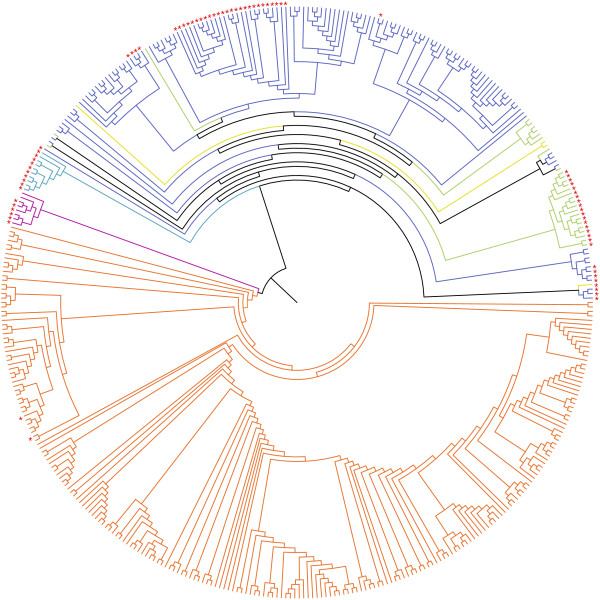
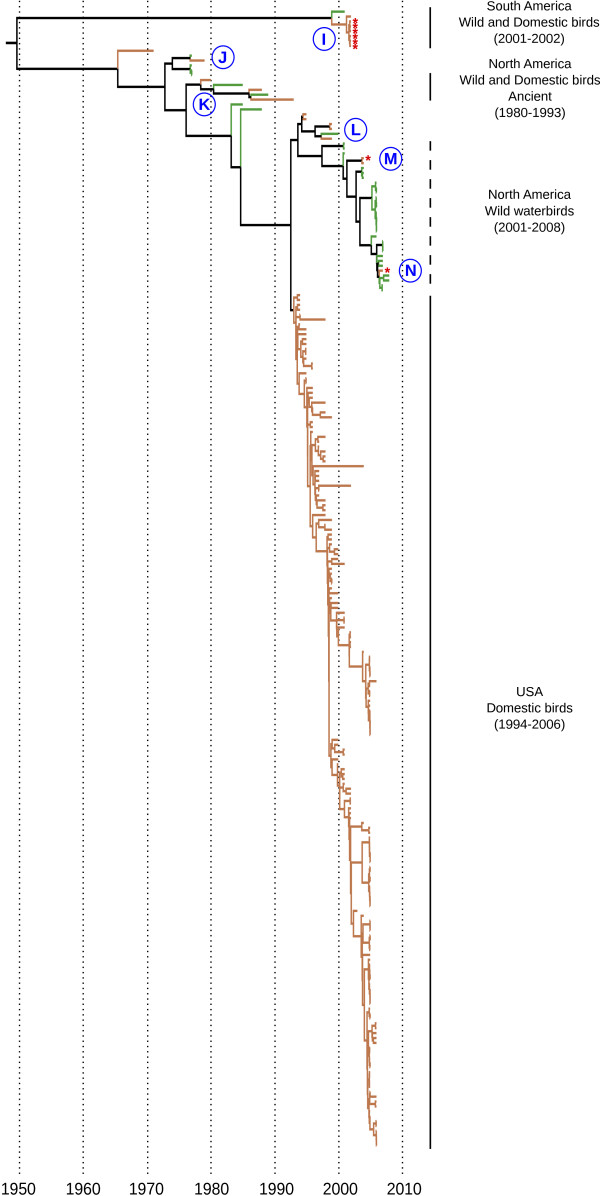
Evolutionary consequences of host shifts represent a challenge to identify the mechanisms involved in the emergence of influenza A (IA) viruses. In this study we focused on the evolutionary history of H7 IA virus in wild and domestic birds, with a particular emphasis on host shifts consequences on the molecular evolution of the hemagglutinin (HA) gene. Based on a dataset of 414 HA nucleotide sequences, we performed an extensive phylogeographic analysis in order to identify the overall genetic structure of H7 IA viruses. We then identified host shift events and investigated viral population dynamics in wild and domestic birds, independently. Finally, we estimated changes in nucleotide substitution rates and tested for positive selection in the HA gene. A strong association between the geographic origin and the genetic structure was observed, with four main clades including viruses isolated in North America, South America, Australia and Eurasia-Africa. We identified ten potential events of virus introduction from wild to domestic birds, but little evidence for spillover of viruses from poultry to wild waterbirds. Several sites involved in host specificity (addition of a glycosylation site in the receptor binding domain) and virulence (insertion of amino acids in the cleavage site) were found to be positively selected in HA nucleotide sequences, in genetically unrelated lineages, suggesting parallel evolution for the HA gene of IA viruses in domestic birds. These results highlight that evolutionary consequences of bird host shifts would need to be further studied to understand the ecological and molecular mechanisms involved in the emergence of domestic bird-adapted viruses.
Figures

References
-
- Barrett R, Kuzawa C, McDade T, Armelagos G. Emerging and re-emerging infectious diseases: the third epidemiological transition. Annu Rev Anthropol. 1998;27:247–271. doi: 10.1146/annurev.anthro.27.1.247. - DOI
-
- Gilbert M, Xiao X, Pfeiffer DU, Epprecht M, Boles S, Czarnecki C, Chaitaweesub P, Kalpravidh W, Minh PQ, Otte MJ, Martin V, Slingenbergh J. Mapping H5N1 highly pathogenic avian influenza risk in Southeast Asia. Proc Natl Acad Sci USA. 2008;105:4769–4774. doi: 10.1073/pnas.0710581105. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
