

Inter-chromosomal variation in the pattern of human population genetic structure
- PMID: 21712187
- PMCID: PMC3326352
- DOI: 10.1186/1479-7364-5-4-220
Inter-chromosomal variation in the pattern of human population genetic structure
Abstract
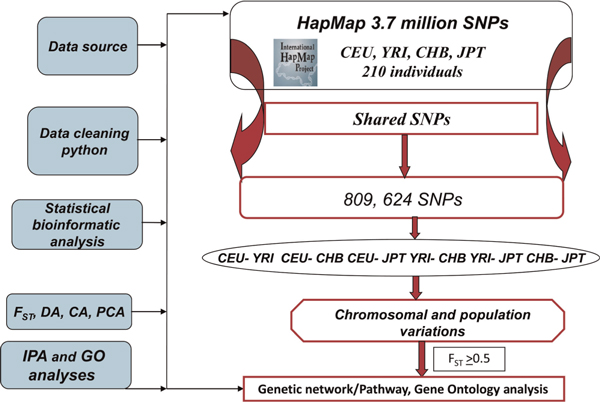
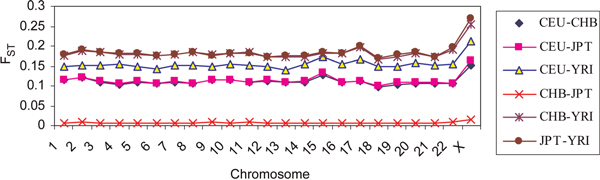
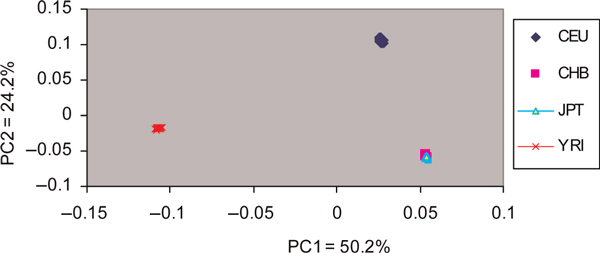
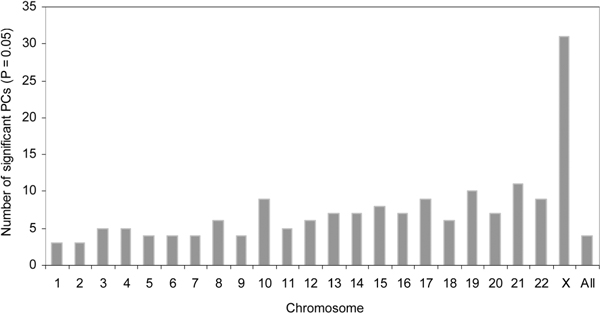
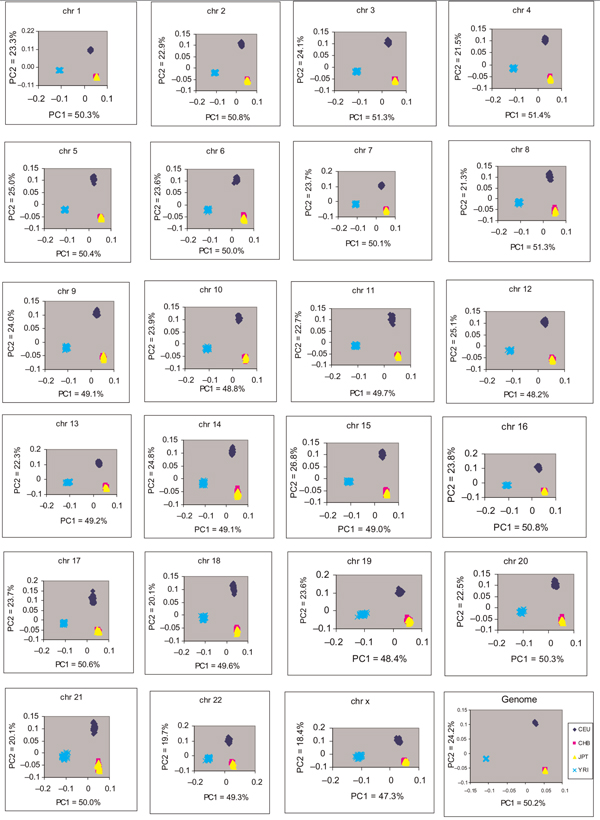
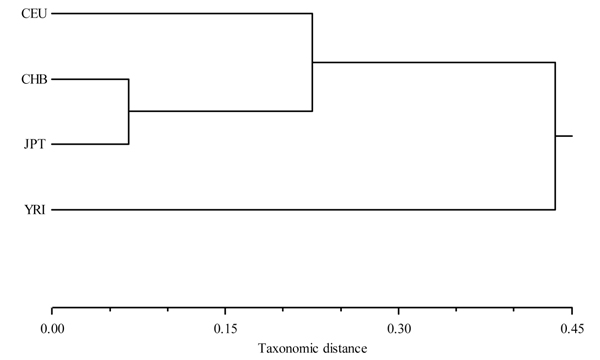
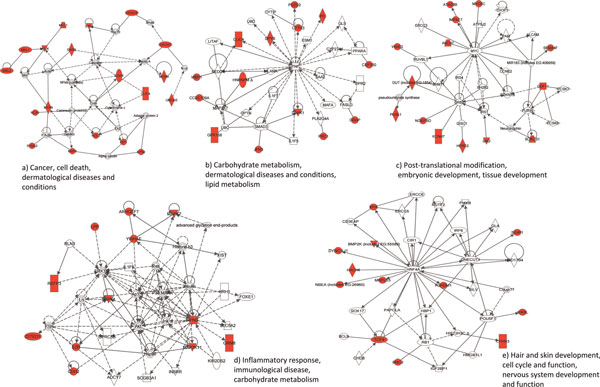
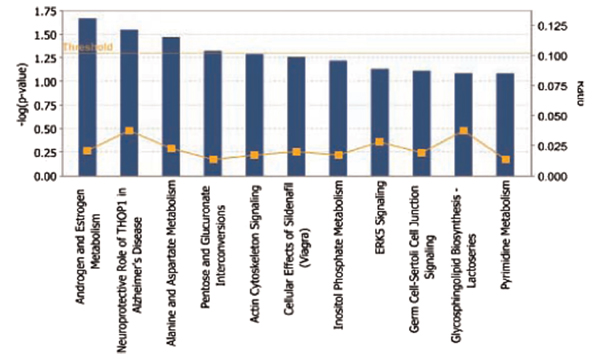
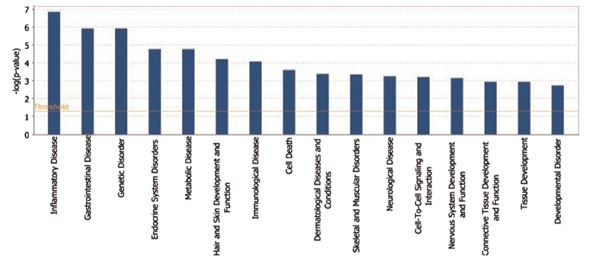
Emerging technologies now make it possible to genotype hundreds of thousands of genetic variations in individuals, across the genome. The study of loci at finer scales will facilitate the understanding of genetic variation at genomic and geographic levels. We examined global and chromosomal variations across HapMap populations using 3.7 million single nucleotide polymorphisms to search for the most stratified genomic regions of human populations and linked these regions to ontological annotation and functional network analysis. To achieve this, we used five complementary statistical and genetic network procedures: principal component (PC), cluster, discriminant, fixation index (FST) and network/pathway analyses. At the global level, the first two PC scores were sufficient to account for major population structure; however, chromosomal level analysis detected subtle forms of population structure within continental populations, and as many as 31 PCs were required to classify individuals into homogeneous groups. Using recommended population ancestry differentiation measures, a total of 126 regions of the genome were catalogued. Gene ontology and networks analyses revealed that these regions included the genes encoding oculocutaneous albinism II (OCA2), hect domain and RLD 2 (HERC2), ectodysplasin A receptor (EDAR) and solute carrier family 45, member 2 (SLC45A2). These genes are associated with melanin production, which is involved in the development of skin and hair colour, skin cancer and eye pigmentation. We also identified the genes encoding interferon-γ (IFNG) and death-associated protein kinase 1 (DAPK1), which are associated with cell death, inflammatory and immunological diseases. An in-depth understanding of these genomic regions may help to explain variations in adaptation to different environments. Our approach offers a comprehensive strategy for analysing chromosome-based population structure and differentiation, and demonstrates the application of complementary statistical and functional network analysis in human genetic variation studies.
Figures
References
-
- Cavalli-Sforza LL, Feldman MW. The application of molecular genetic approaches to the study of human evolution. Nat Genet. 2003;33(Suppl):266–275. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
