

Unraveling the scissile bond: how ADAMTS13 recognizes and cleaves von Willebrand factor
- PMID: 21715306
- PMCID: PMC3179391
- DOI: 10.1182/blood-2011-02-306597
Unraveling the scissile bond: how ADAMTS13 recognizes and cleaves von Willebrand factor
Abstract
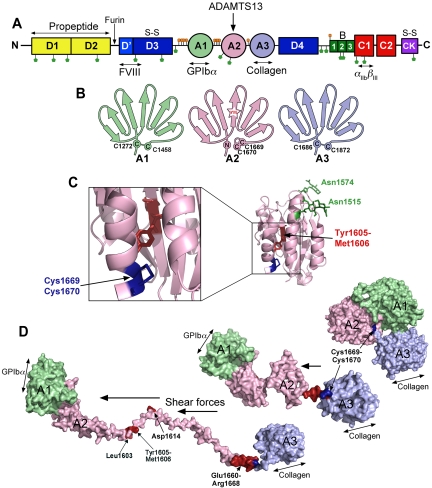
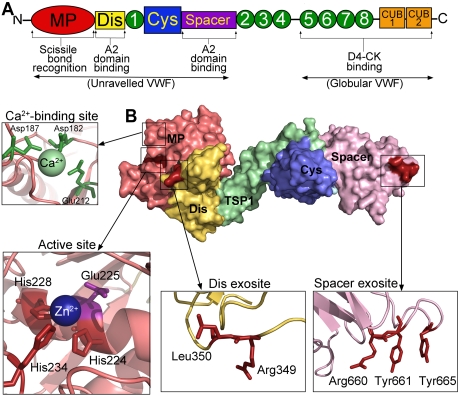
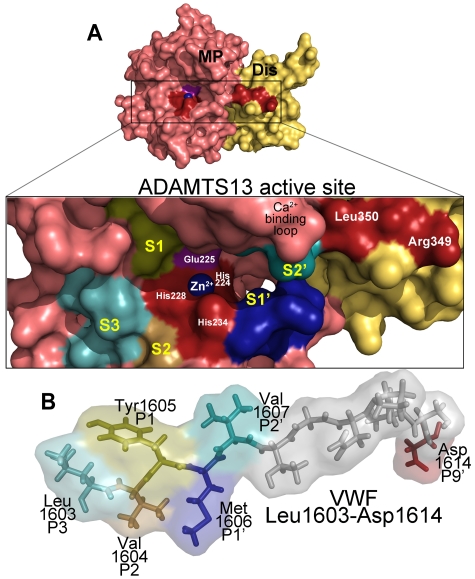
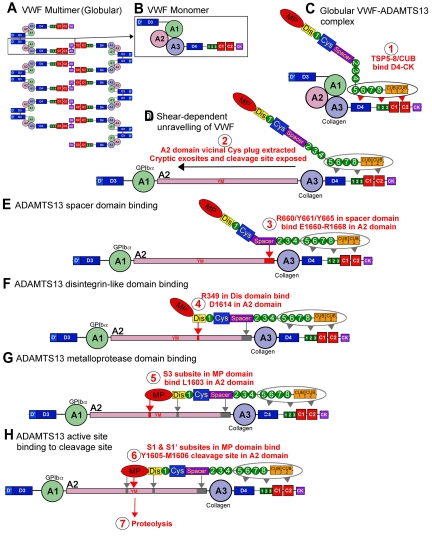
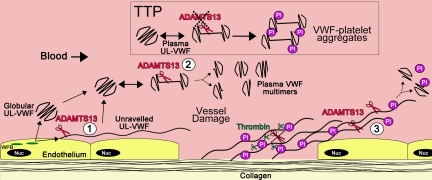
von Willebrand factor (VWF) is a large adhesive glycoprotein with established functions in hemostasis. It serves as a carrier for factor VIII and acts as a vascular damage sensor by attracting platelets to sites of vessel injury. VWF size is important for this latter function, with larger multimers being more hemostatically active. Functional imbalance in multimer size can variously cause microvascular thrombosis or bleeding. The regulation of VWF multimeric size and platelet-tethering function is carried out by ADAMTS13, a plasma metalloprotease that is constitutively active. Unusually, protease activity of ADAMTS13 is controlled not by natural inhibitors but by conformational changes in its substrate, which are induced when VWF is subject to elevated rheologic shear forces. This transforms VWF from a globular to an elongated protein. This conformational transformation unfolds the VWF A2 domain and reveals cryptic exosites as well as the scissile bond. To enable VWF proteolysis, ADAMTS13 makes multiple interactions that bring the protease to the substrate and position it to engage with the cleavage site as this becomes exposed by shear. This article reviews recent literature on the interaction between these 2 multidomain proteins and provides a summary model to explain proteolytic regulation of VWF by ADAMTS13.
Figures
References
-
- Sadler JE. Biochemistry and genetics of von Willebrand factor. Annu Rev Biochem. 1998;67:395–424. - PubMed
-
- Roth GJ, Titani K, Hoyer LW, Hickey MJ. Localization of binding sites within human von Willebrand factor for monomeric type III collagen. Biochemistry. 1986;25(26):8357–8361. - PubMed
-
- Hamer RJ, Koedam JA, Beeser-Visser NH, Bertina RM, Van Mourik JA, Sixma JJ. Factor VIII binds to von Willebrand factor via its Mr-80,000 light chain. Eur J Biochem. 1987;166(1):37–43. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
