

Improving pan-genome annotation using whole genome multiple alignment
- PMID: 21718539
- PMCID: PMC3142524
- DOI: 10.1186/1471-2105-12-272
Improving pan-genome annotation using whole genome multiple alignment
Abstract
Background: Rapid annotation and comparisons of genomes from multiple isolates (pan-genomes) is becoming commonplace due to advances in sequencing technology. Genome annotations can contain inconsistencies and errors that hinder comparative analysis even within a single species. Tools are needed to compare and improve annotation quality across sets of closely related genomes.
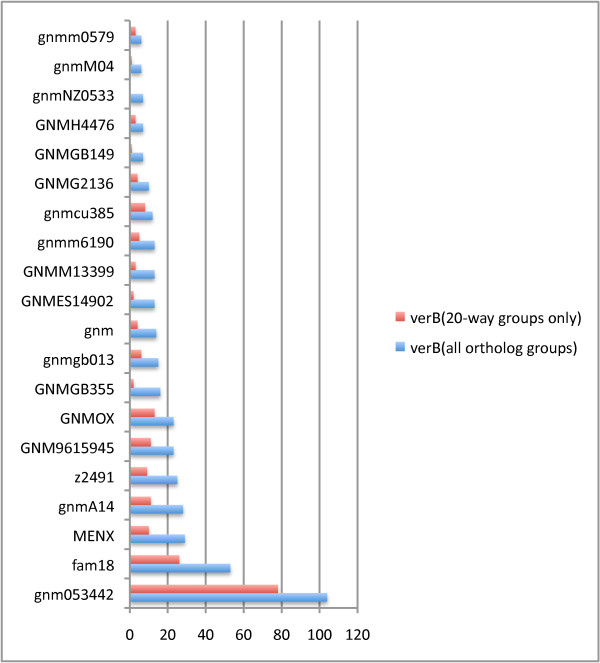
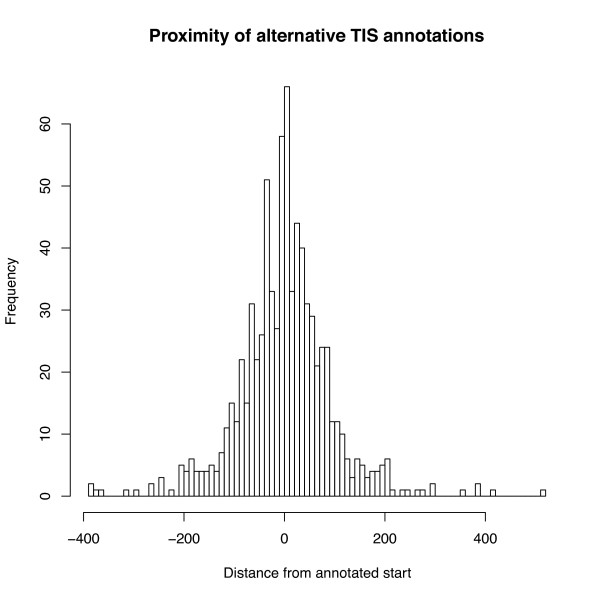
Results: We introduce a new tool, Mugsy-Annotator, that identifies orthologs and evaluates annotation quality in prokaryotic genomes using whole genome multiple alignment. Mugsy-Annotator identifies anomalies in annotated gene structures, including inconsistently located translation initiation sites and disrupted genes due to draft genome sequencing or pseudogenes. An evaluation of species pan-genomes using the tool indicates that such anomalies are common, especially at translation initiation sites. Mugsy-Annotator reports alternate annotations that improve consistency and are candidates for further review.
Conclusions: Whole genome multiple alignment can be used to efficiently identify orthologs and annotation problem areas in a bacterial pan-genome. Comparisons of annotated gene structures within a species may show more variation than is actually present in the genome, indicating errors in genome annotation. Our new tool Mugsy-Annotator assists re-annotation efforts by highlighting edits that improve annotation consistency.
© 2011 Angiuoli et al; licensee BioMed Central Ltd.
Figures





References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
