

Dravet syndrome as epileptic encephalopathy: evidence from long-term course and neuropathology
- PMID: 21719429
- PMCID: PMC3187538
- DOI: 10.1093/brain/awr129
Dravet syndrome as epileptic encephalopathy: evidence from long-term course and neuropathology
Abstract
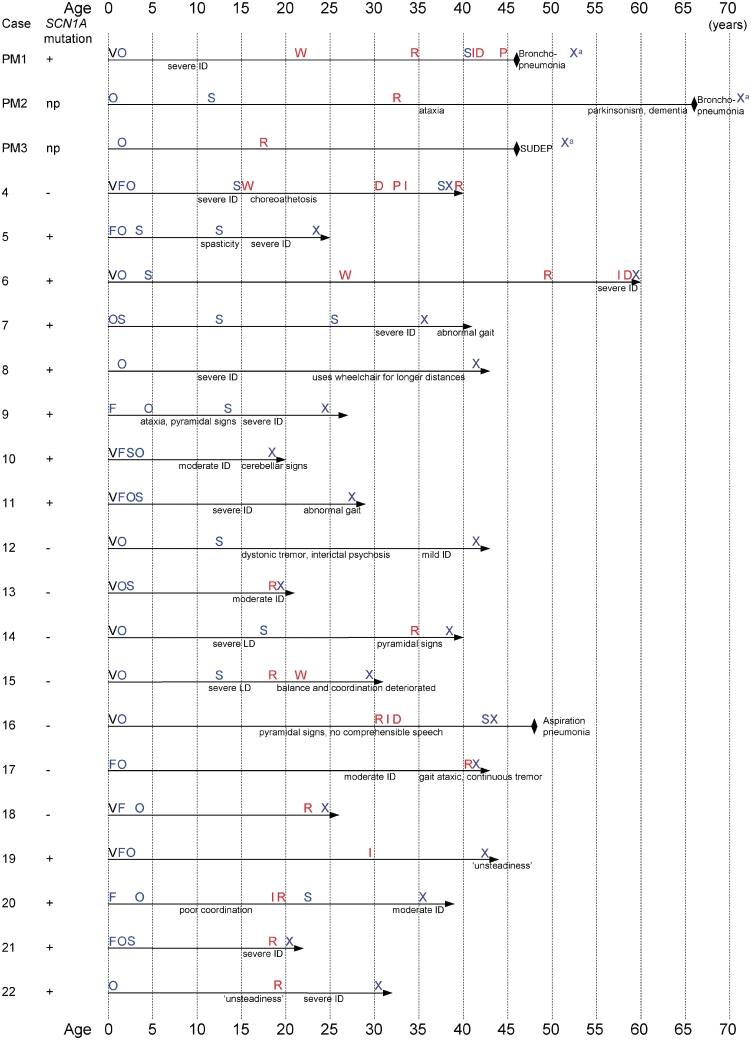
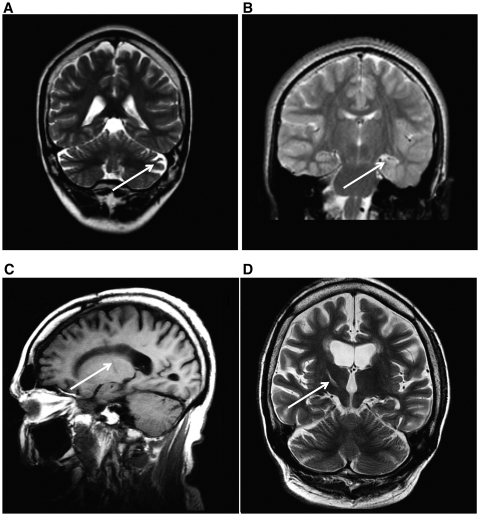
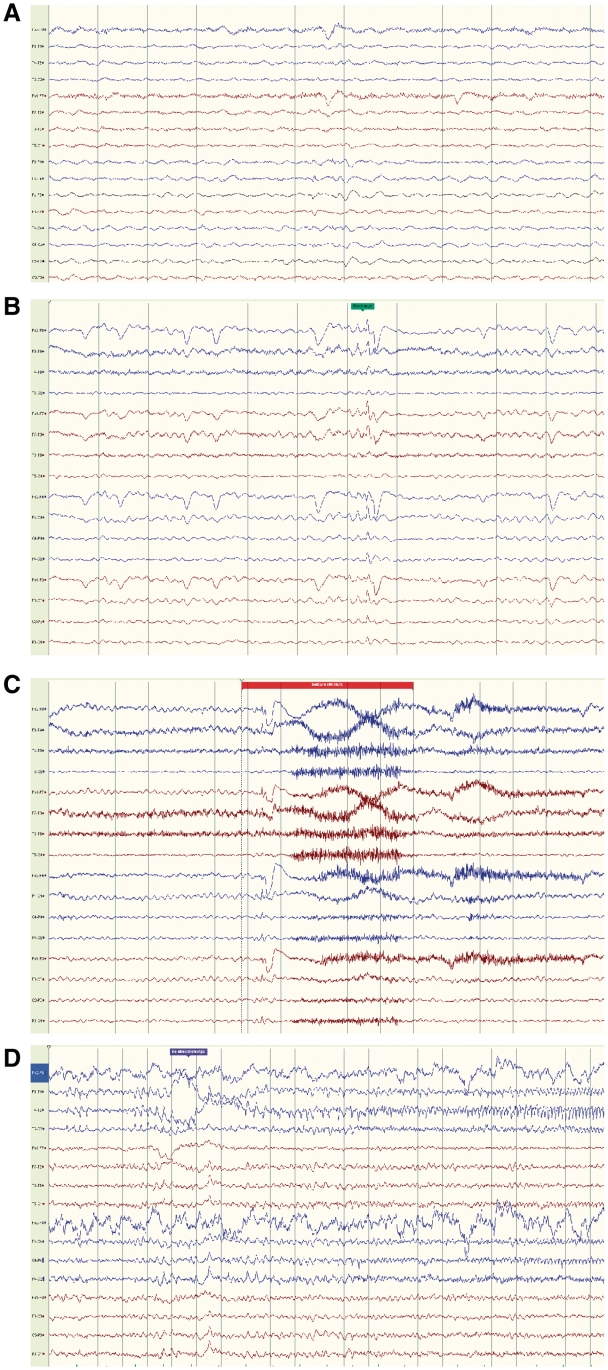
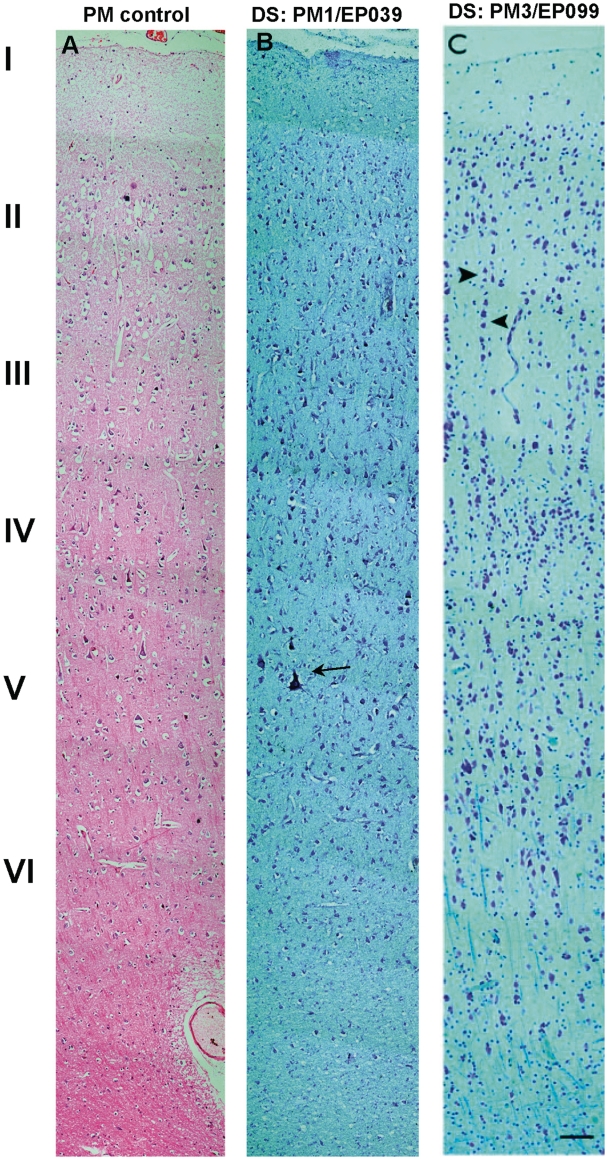
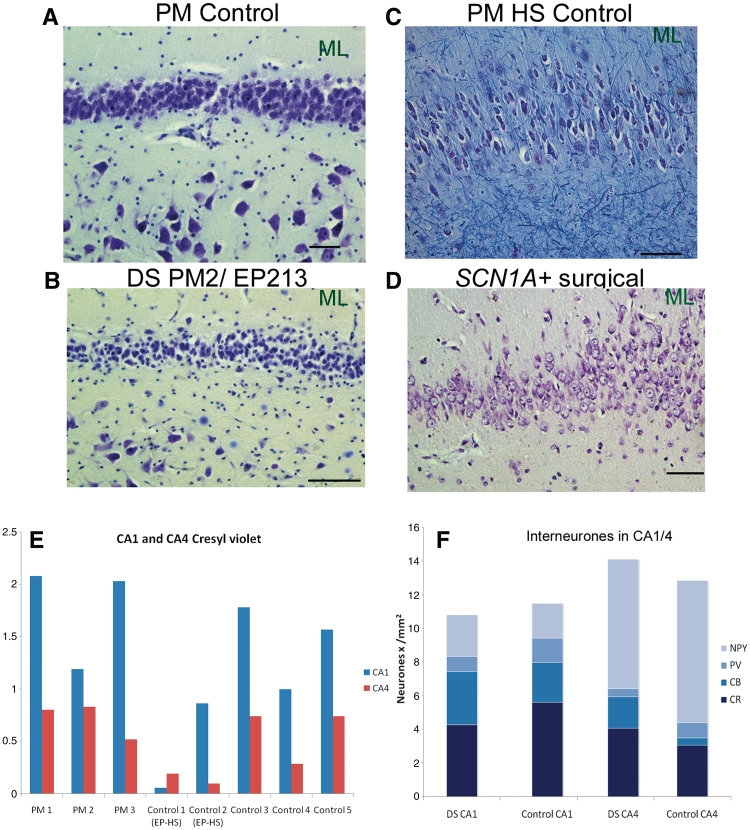
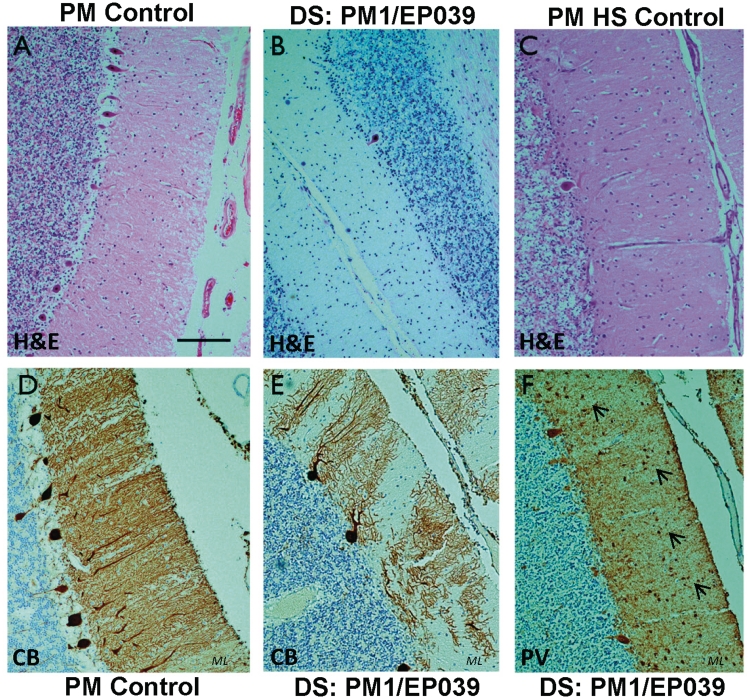
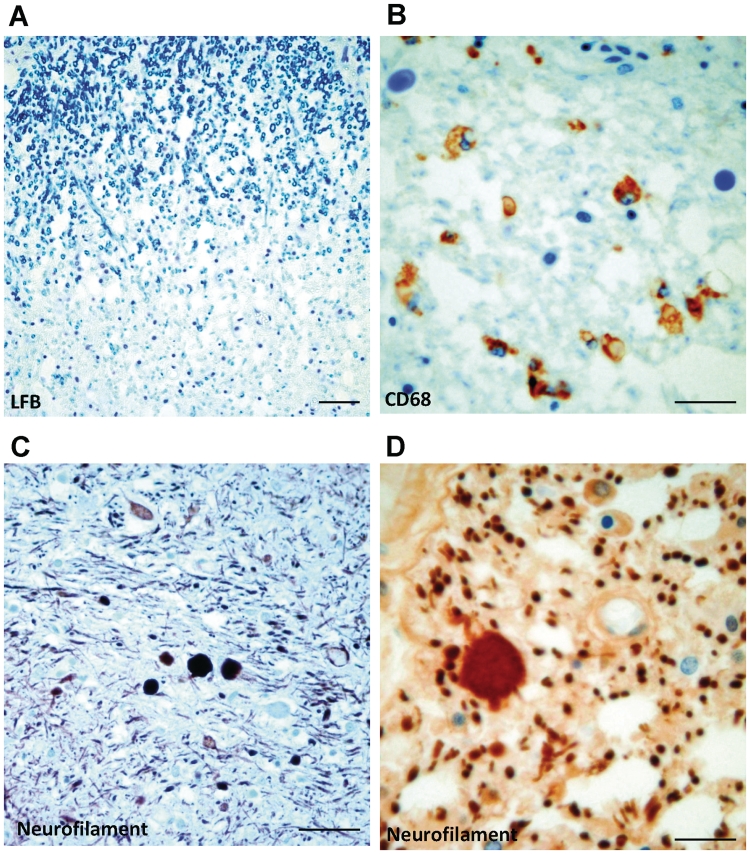
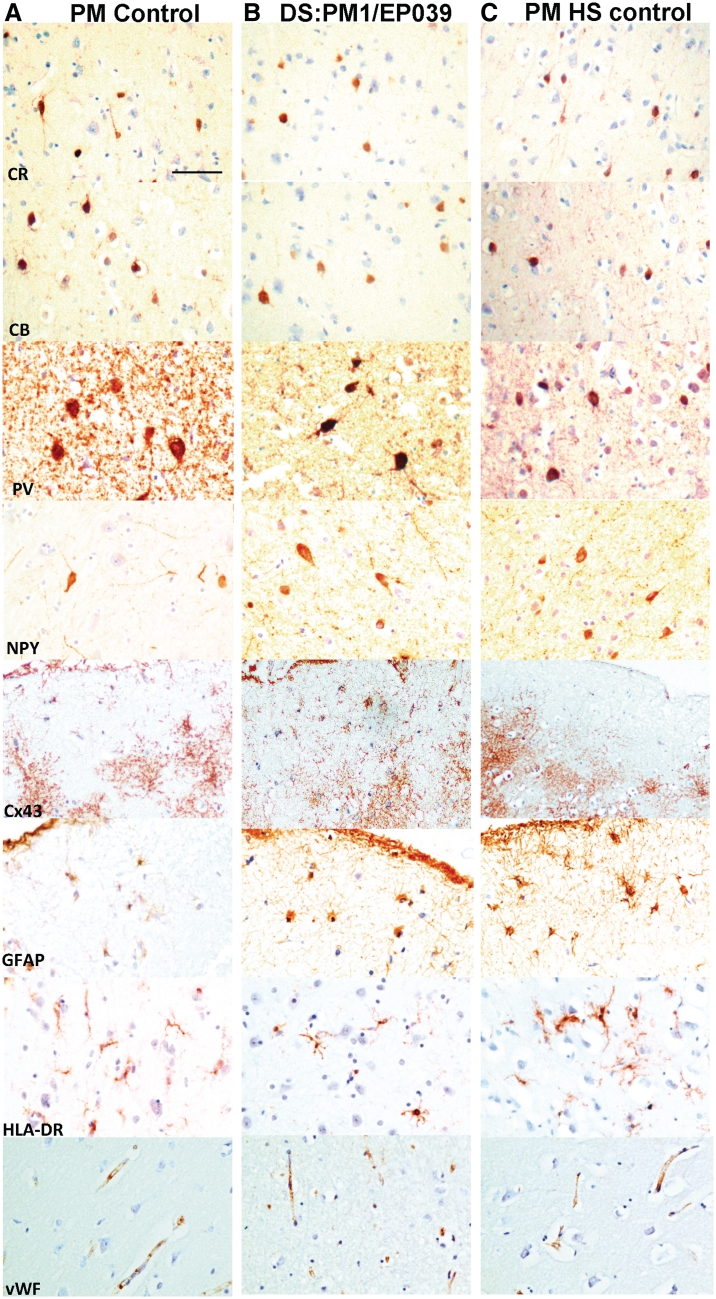
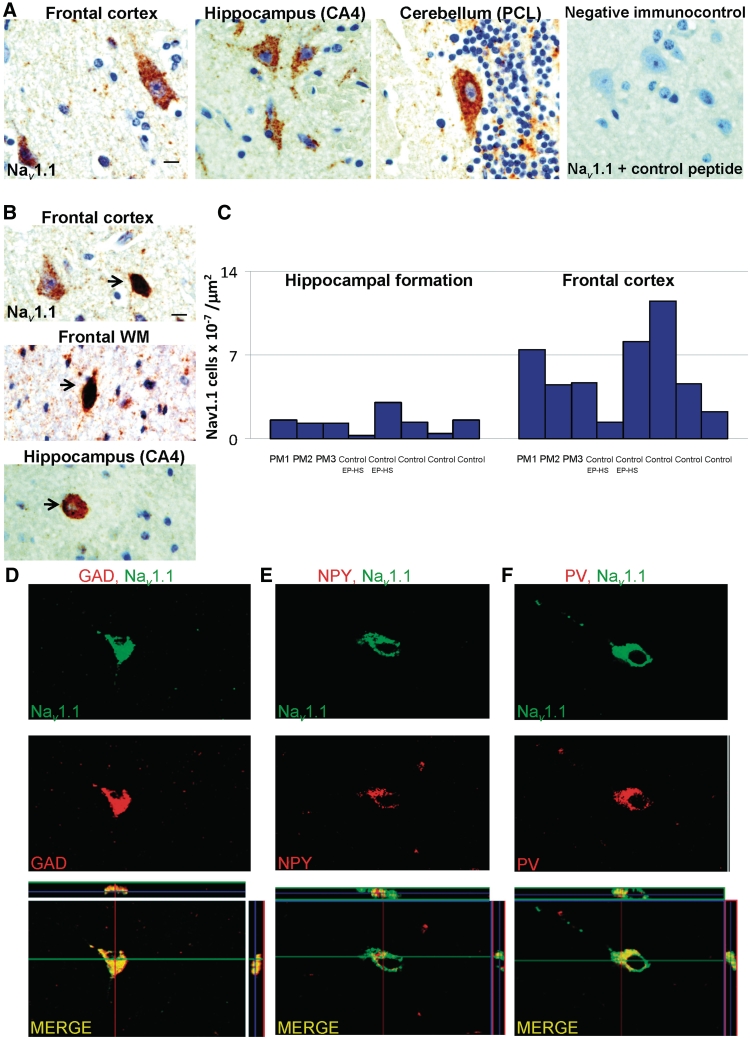
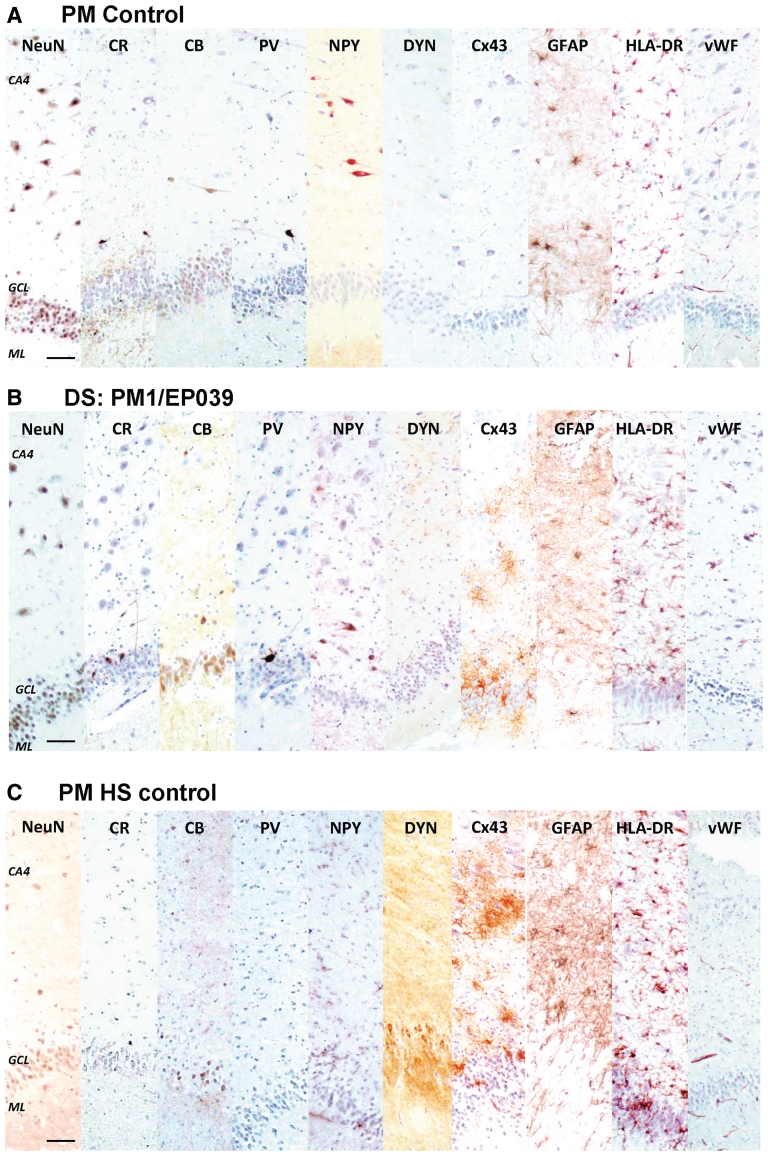
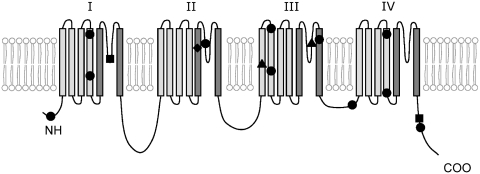
Dravet syndrome is an epilepsy syndrome of infantile onset, frequently caused by SCN1A mutations or deletions. Its prevalence, long-term evolution in adults and neuropathology are not well known. We identified a series of 22 adult patients, including three adult post-mortem cases with Dravet syndrome. For all patients, we reviewed the clinical history, seizure types and frequency, antiepileptic drugs, cognitive, social and functional outcome and results of investigations. A systematic neuropathology study was performed, with post-mortem material from three adult cases with Dravet syndrome, in comparison with controls and a range of relevant paediatric tissue. Twenty-two adults with Dravet syndrome, 10 female, were included, median age 39 years (range 20-66). SCN1A structural variation was found in 60% of the adult Dravet patients tested, including one post-mortem case with DNA extracted from brain tissue. Novel mutations were described for 11 adult patients; one patient had three SCN1A mutations. Features of Dravet syndrome in adulthood include multiple seizure types despite polytherapy, and age-dependent evolution in seizure semiology and electroencephalographic pattern. Fever sensitivity persisted through adulthood in 11 cases. Neurological decline occurred in adulthood with cognitive and motor deterioration. Dysphagia may develop in or after the fourth decade of life, leading to significant morbidity, or death. The correct diagnosis at an older age made an impact at several levels. Treatment changes improved seizure control even after years of drug resistance in all three cases with sufficient follow-up after drug changes were instituted; better control led to significant improvement in cognitive performance and quality of life in adulthood in two cases. There was no histopathological hallmark feature of Dravet syndrome in this series. Strikingly, there was remarkable preservation of neurons and interneurons in the neocortex and hippocampi of Dravet adult post-mortem cases. Our study provides evidence that Dravet syndrome is at least in part an epileptic encephalopathy.
Figures
References
-
- Akiyama M, Kobayashi K, Yoshinaga H, Ohtsuka Y. A long-term follow-up study of Dravet syndrome up to adulthood. Epilepsia. 2010;51:1043–52. - PubMed
-
- Auvin S, Dulac O, Vallée L. Do SCN1A mutations protect from hippocampal sclerosis? Epilepsia. 2008;49:1107–8. - PubMed
-
- Baulac S, Gourfinkel-An I, Nabbout R, Huberfeld G, Serratosa J, Leguern E, et al. Fever, genes, and epilepsy. Lancet Neurol. 2004;3:421–30. - PubMed
-
- Berg AT, Berkovic SF, Brodie MJ, Buchhalter J, Cross JH, van Emde Boas W, et al. Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE Commission on Classification and Terminology, 2005-2009. Epilepsia. 2010;51:676–85. - PubMed
-
- Berkovic SF, Harkin L, McMahon JM, Pelekanos JT, Zuberi SM, Wirrell EC, et al. De novo mutations of the sodium channel gene SCN1A in alleged vaccine encephalopathy: a retrospective study. Lancet Neurol. 2006;5:488–92. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
