

Clarifying lysosomal storage diseases
- PMID: 21723623
- PMCID: PMC3153126
- DOI: 10.1016/j.tins.2011.05.006
Clarifying lysosomal storage diseases
Abstract
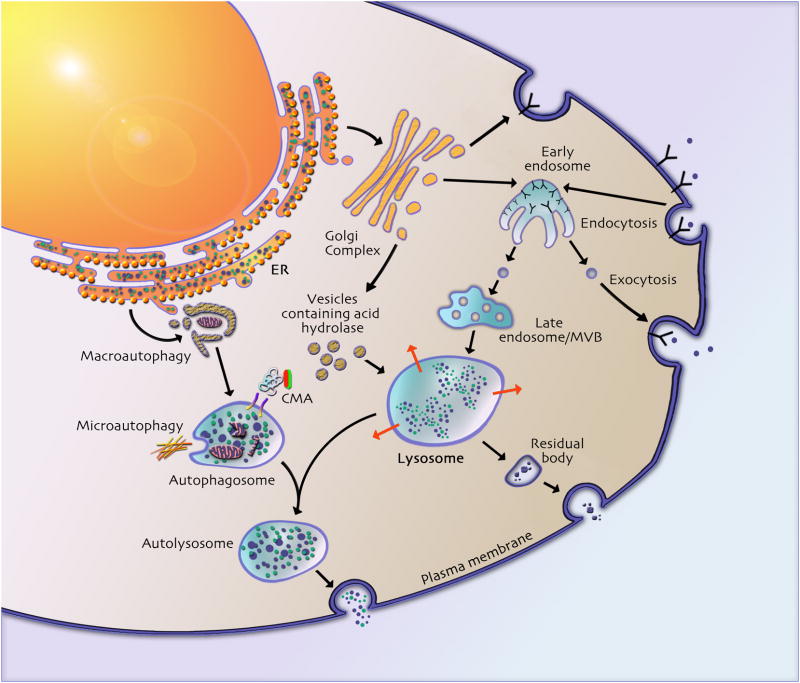
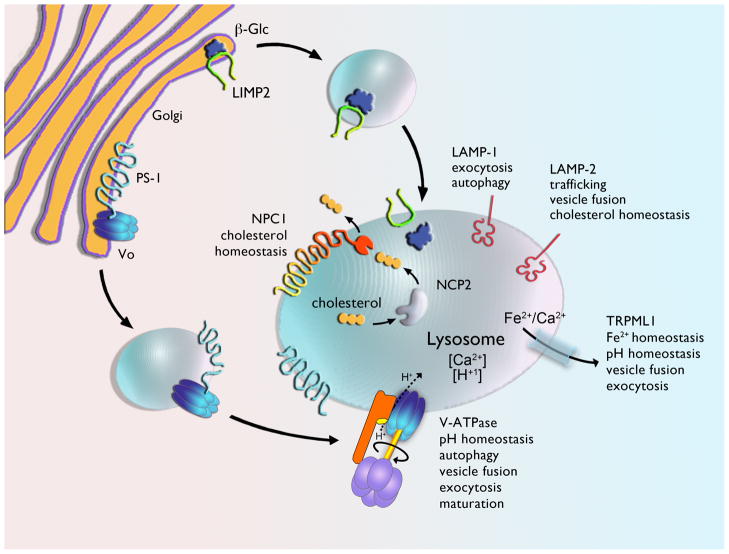
Lysosomal storage diseases (LSDs) are a class of metabolic disorders caused by mutations in proteins critical for lysosomal function. Such proteins include lysosomal enzymes, lysosomal integral membrane proteins, and proteins involved in the post-translational modification and trafficking of lysosomal proteins. There are many recognized forms of LSDs and, although individually rare, their combined prevalence is estimated to be 1 in 8000 births. Over two-thirds of LSDs involve central nervous system (CNS) dysfunction (progressive cognitive and motor decline) and these symptoms are often the most debilitating. Although the genetic basis for these disorders is clear and the biochemistry of the proteins well understood, the cellular mechanisms by which deficiencies in these proteins disrupt neuronal viability remain ambiguous. In this review, we provide an overview of the widespread cellular perturbations occurring in LSDs, how they might be linked and interventions that may specifically or globally correct those defects.
Copyright © 2011 Elsevier Ltd. All rights reserved.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
