

Enzyme inhibition by allosteric capture of an inactive conformation
- PMID: 21723875
- PMCID: PMC3157250
- DOI: 10.1016/j.jmb.2011.06.032
Enzyme inhibition by allosteric capture of an inactive conformation
Abstract
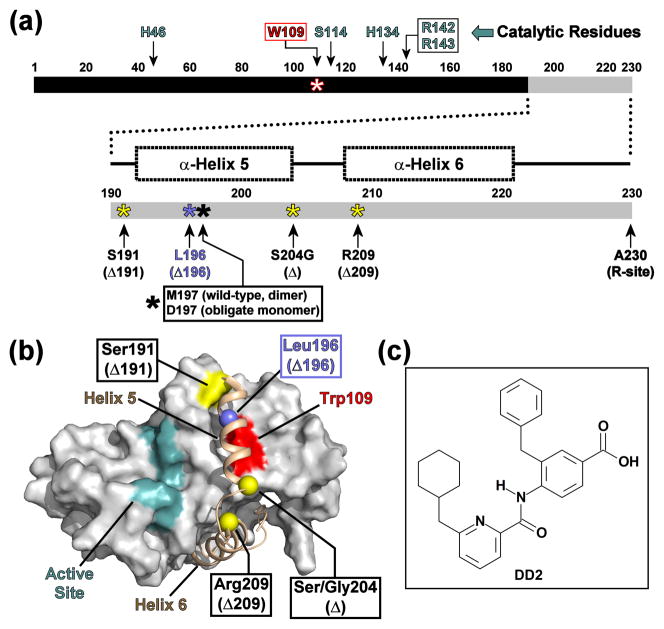
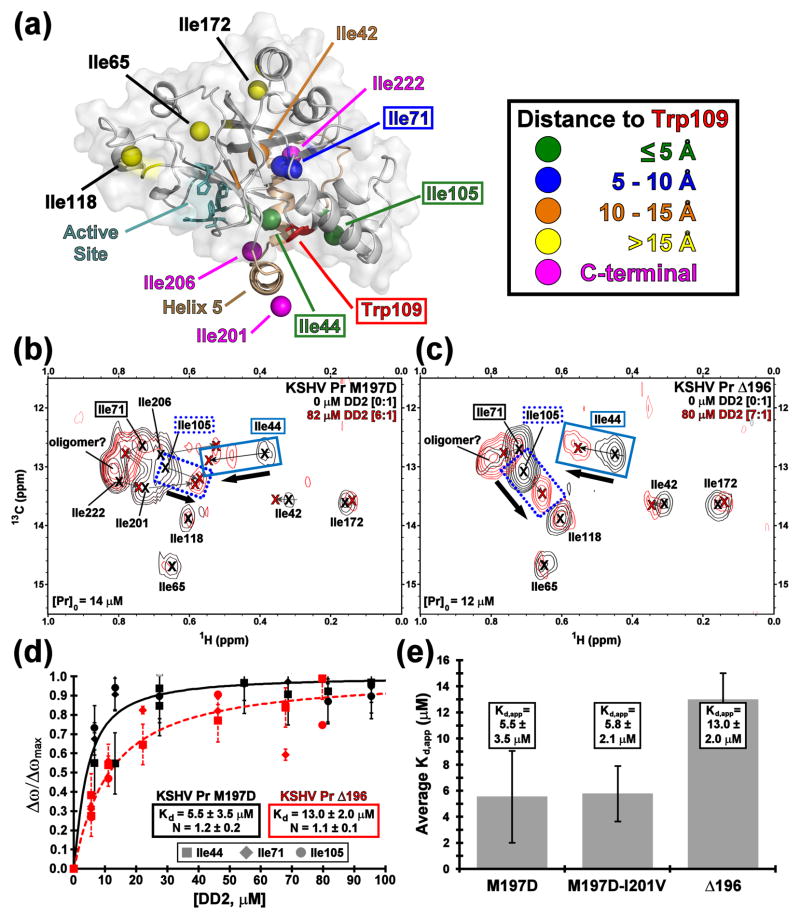
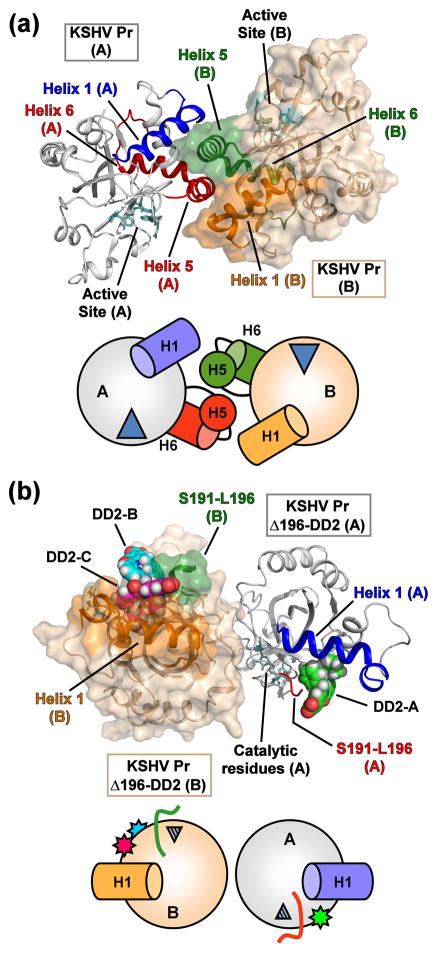
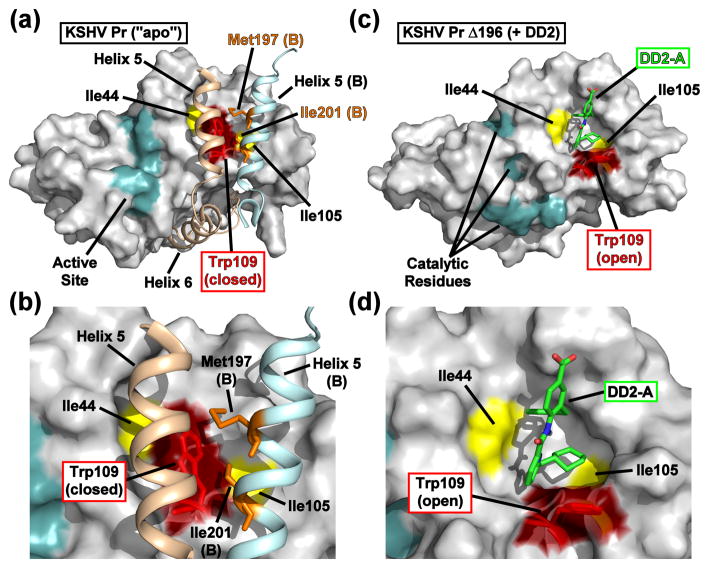
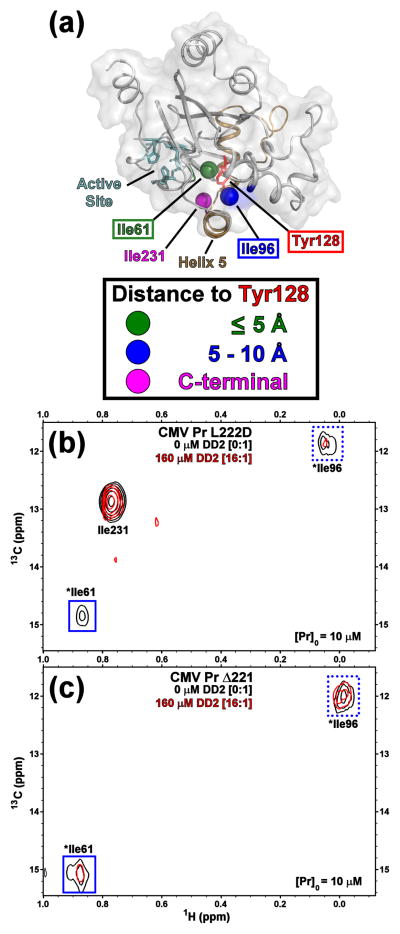
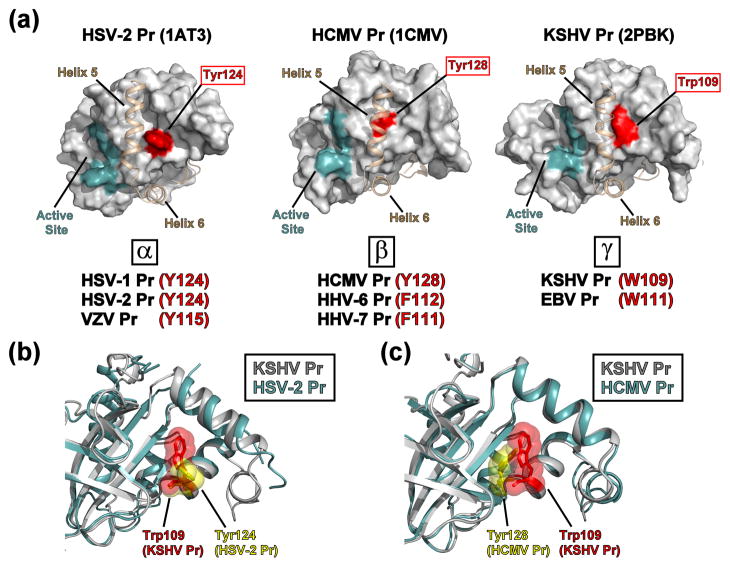
All members of the human herpesvirus protease (HHV Pr) family are active as weakly associating dimers but inactive as monomers. A small-molecule allosteric inhibitor of Kaposi's sarcoma-associated herpesvirus protease (KSHV Pr) traps the enzyme in an inactive monomeric state where the C-terminal helices are unfolded and the hydrophobic dimer interface is exposed. NMR titration studies demonstrate that the inhibitor binds to KSHV Pr monomers with low micromolar affinity. A 2.0-Å-resolution X-ray crystal structure of a C-terminal truncated KSHV Pr-inhibitor complex locates the binding pocket at the dimer interface and displays significant conformational perturbations at the active site, 15 Å from the allosteric site. NMR and CD data suggest that the small molecule inhibits human cytomegalovirus protease via a similar mechanism. As all HHV Prs are functionally and structurally homologous, the inhibitor represents a class of compounds that may be developed into broad-spectrum therapeutics that allosterically regulate enzymatic activity by disrupting protein-protein interactions.
Copyright © 2011 Elsevier Ltd. All rights reserved.
Figures
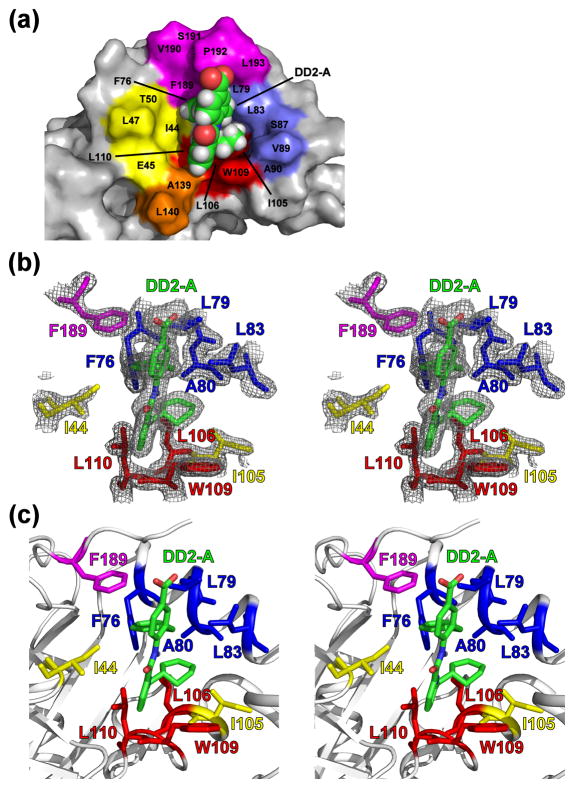
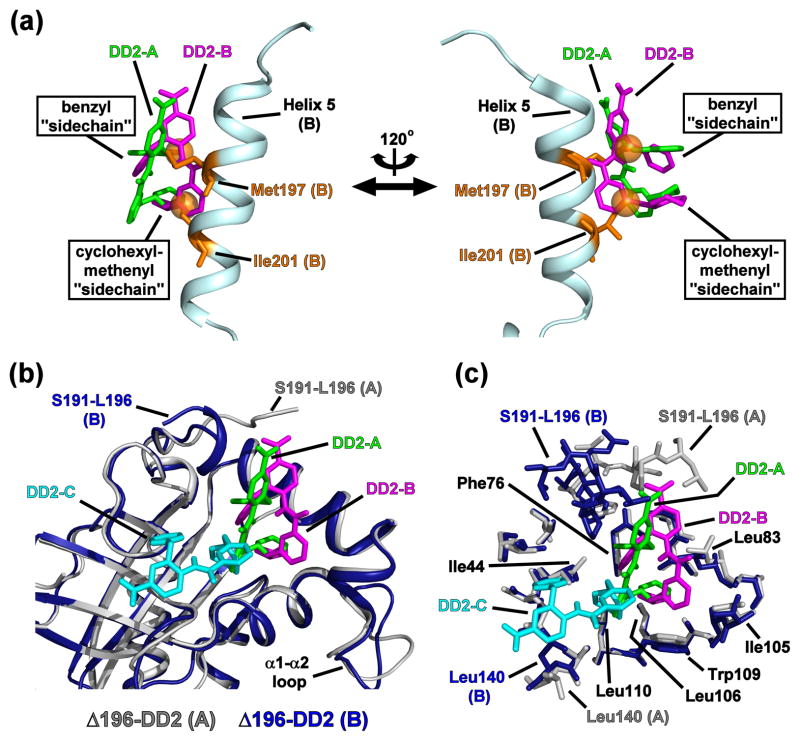
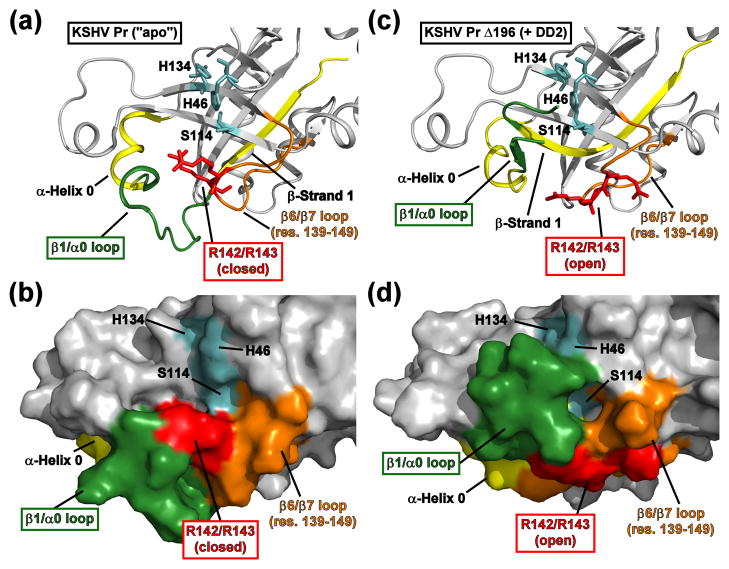
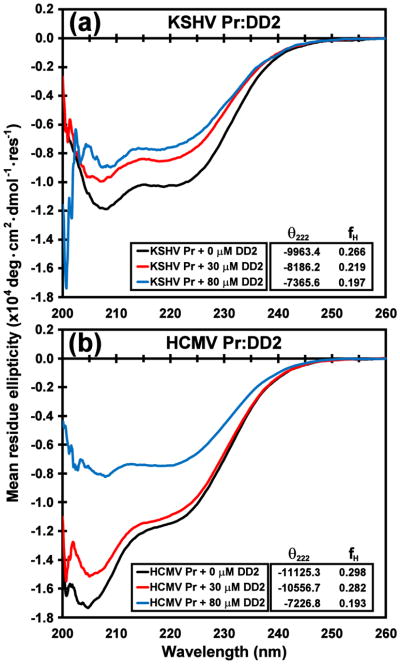
References
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
