

Prolactin-stimulated activation of ERK1/2 mitogen-activated protein kinases is controlled by PI3-kinase/Rac/PAK signaling pathway in breast cancer cells
- PMID: 21726627
- PMCID: PMC3156300
- DOI: 10.1016/j.cellsig.2011.06.014
Prolactin-stimulated activation of ERK1/2 mitogen-activated protein kinases is controlled by PI3-kinase/Rac/PAK signaling pathway in breast cancer cells
Abstract
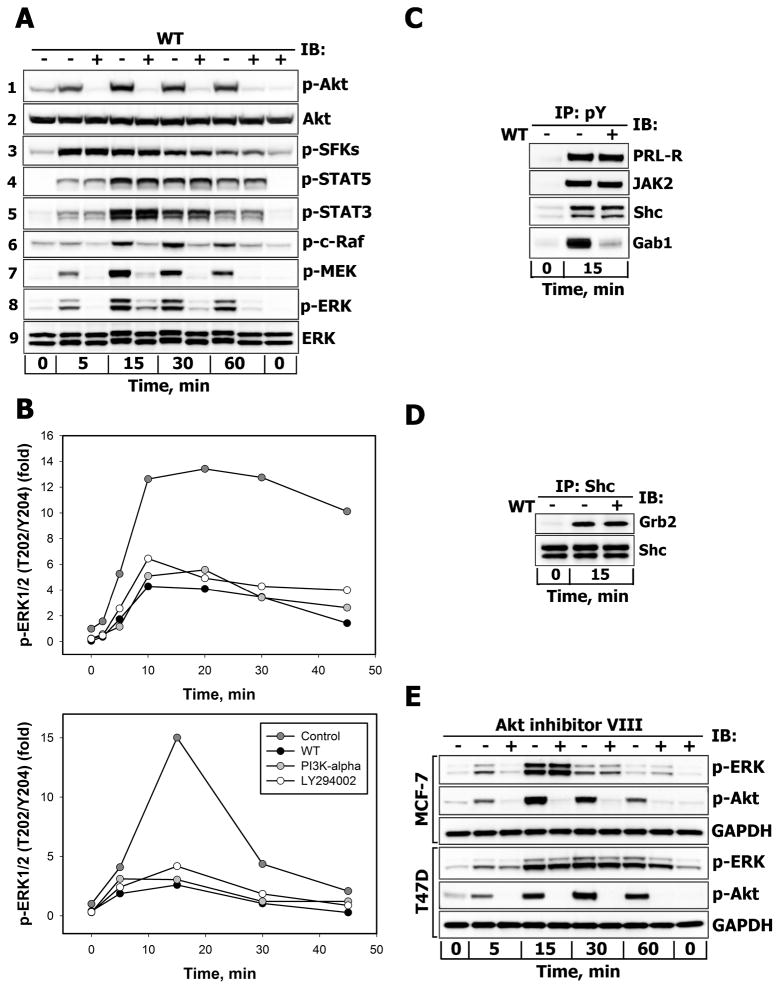
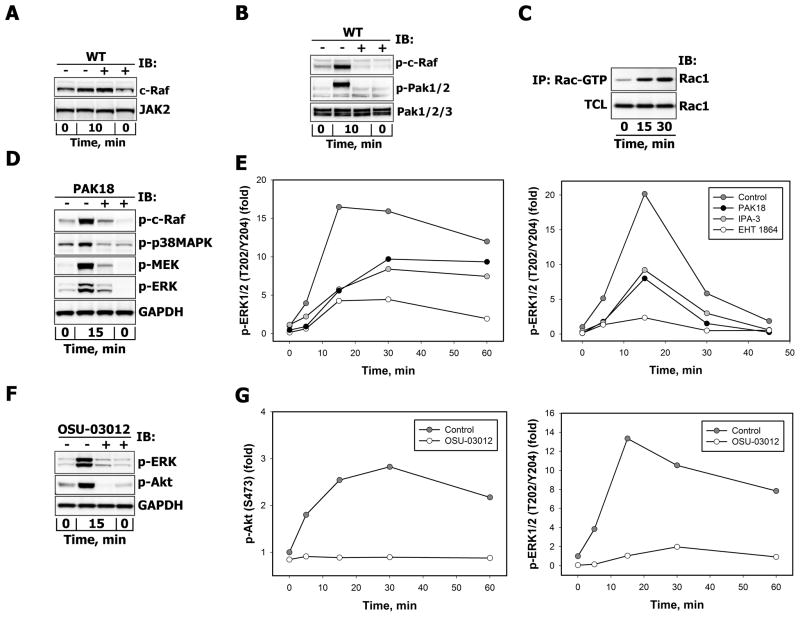
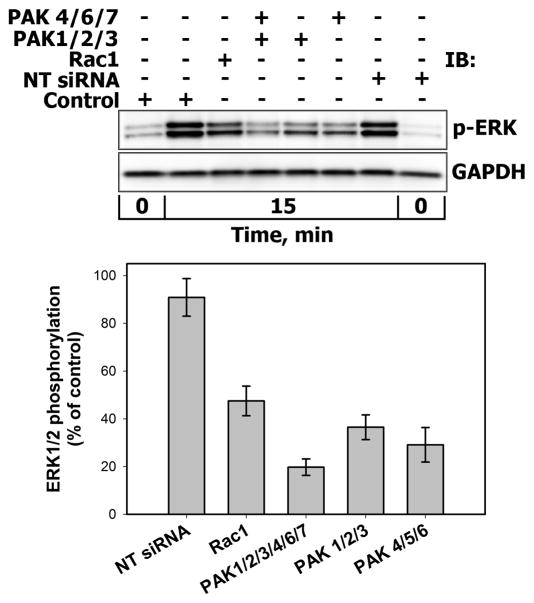
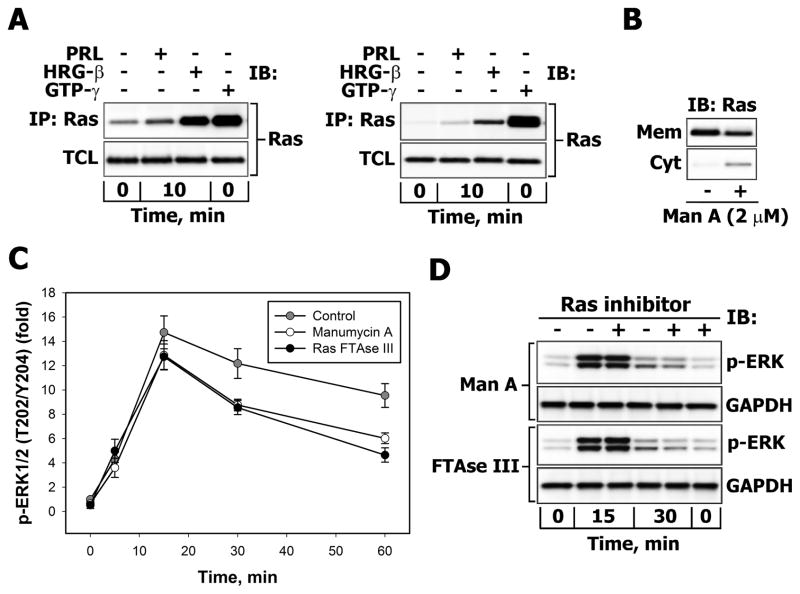
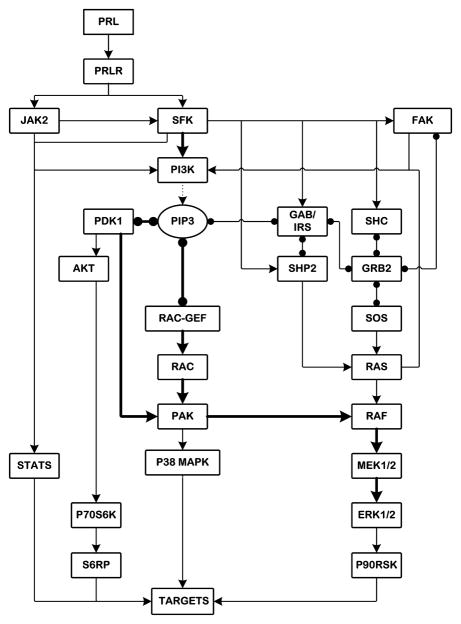
There is strong evidence that deregulation of prolactin (PRL) signaling contributes to pathogenesis and chemoresistance of breast cancer. Therefore, understanding cross-talk between distinct signal transduction pathways triggered by activation of the prolactin receptor (PRL-R), is essential for elucidating the pathogenesis of metastatic breast cancer. In this study, we applied a sequential inhibitory analysis of various signaling intermediates to examine the hierarchy of protein interactions within the PRL signaling network and to evaluate the relative contributions of multiple signaling branches downstream of PRL-R to the activation of the extracellular signal-regulated kinases ERK1 and ERK2 in T47D and MCF-7 human breast cancer cells. Quantitative measurements of the phosphorylation/activation patterns of proteins showed that PRL simultaneously activated Src family kinases (SFKs) and the JAK/STAT, phosphoinositide-3 (PI3)-kinase/Akt and MAPK signaling pathways. The specific blockade or siRNA-mediated suppression of SFK/FAK, JAK2/STAT5, PI3-kinase/PDK1/Akt, Rac/PAK or Ras regulatory circuits revealed that (1) the PI3-kinase/Akt pathway is required for activation of the MAPK/ERK signaling cascade upon PRL stimulation; (2) PI3-kinase-mediated activation of the c-Raf-MEK1/2-ERK1/2 cascade occurs independent of signaling dowstream of STATs, Akt and PKC, but requires JAK2, SFKs and FAK activities; (3) activated PRL-R mainly utilizes the PI3-kinase-dependent Rac/PAK pathway rather than the canonical Shc/Grb2/SOS/Ras route to initiate and sustain ERK1/2 signaling. By interconnecting diverse signaling pathways PLR may enhance proliferation, survival, migration and invasiveness of breast cancer cells.
Copyright © 2011 Elsevier Inc. All rights reserved.
Figures
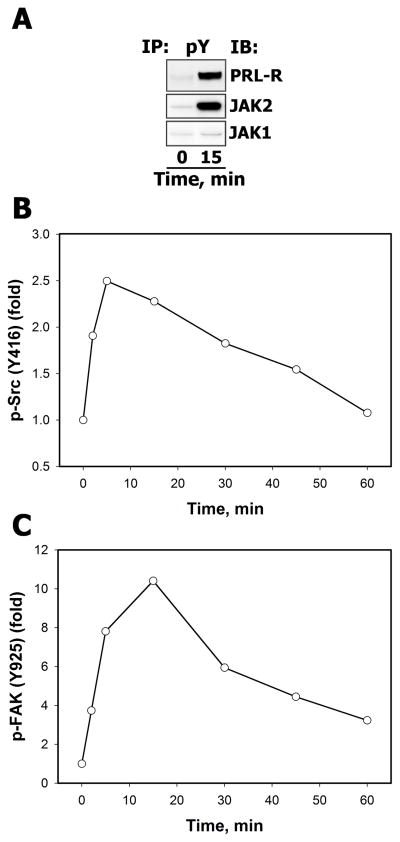
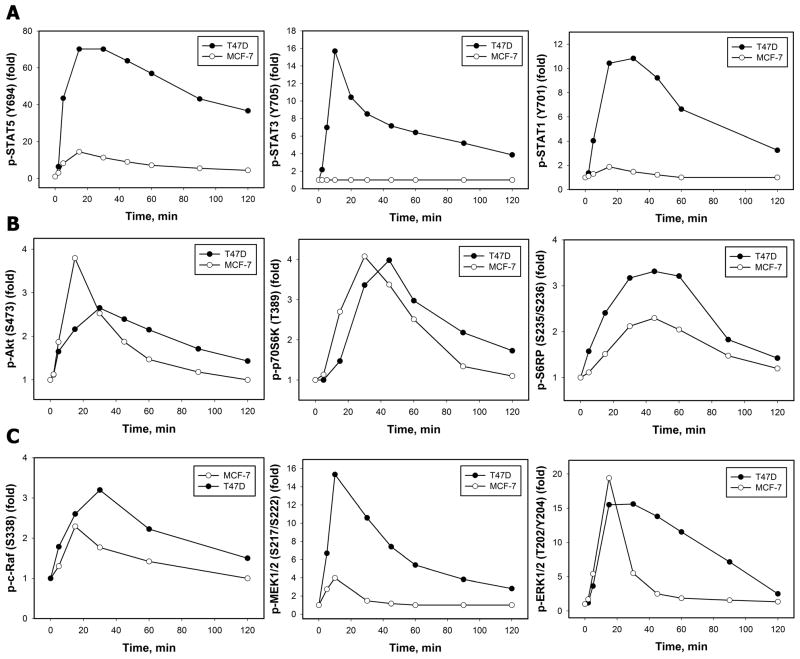
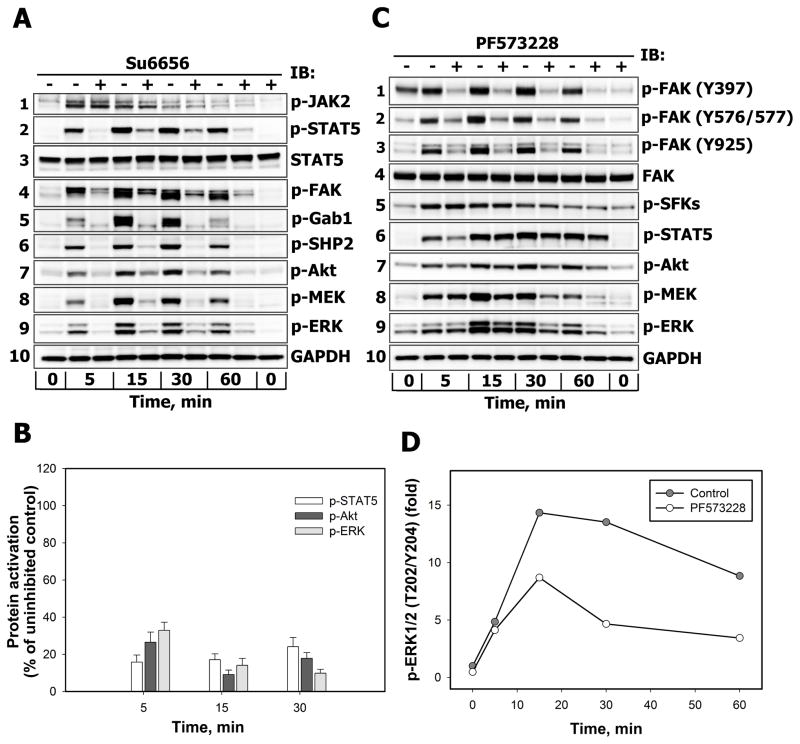
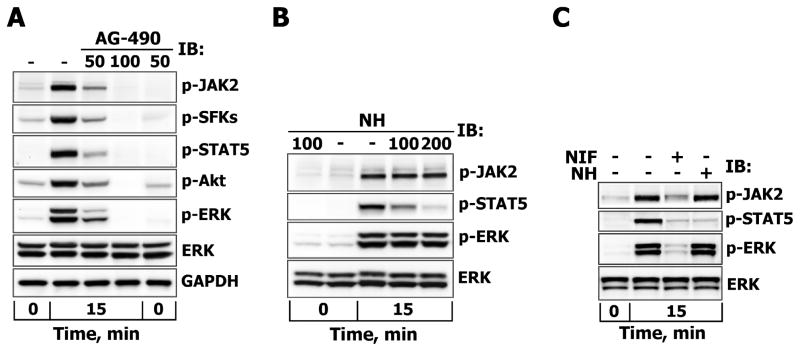
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
