

Opposing effects of glutamine and asparagine govern prion formation by intrinsically disordered proteins
- PMID: 21726811
- PMCID: PMC3132398
- DOI: 10.1016/j.molcel.2011.05.013
Opposing effects of glutamine and asparagine govern prion formation by intrinsically disordered proteins
Abstract
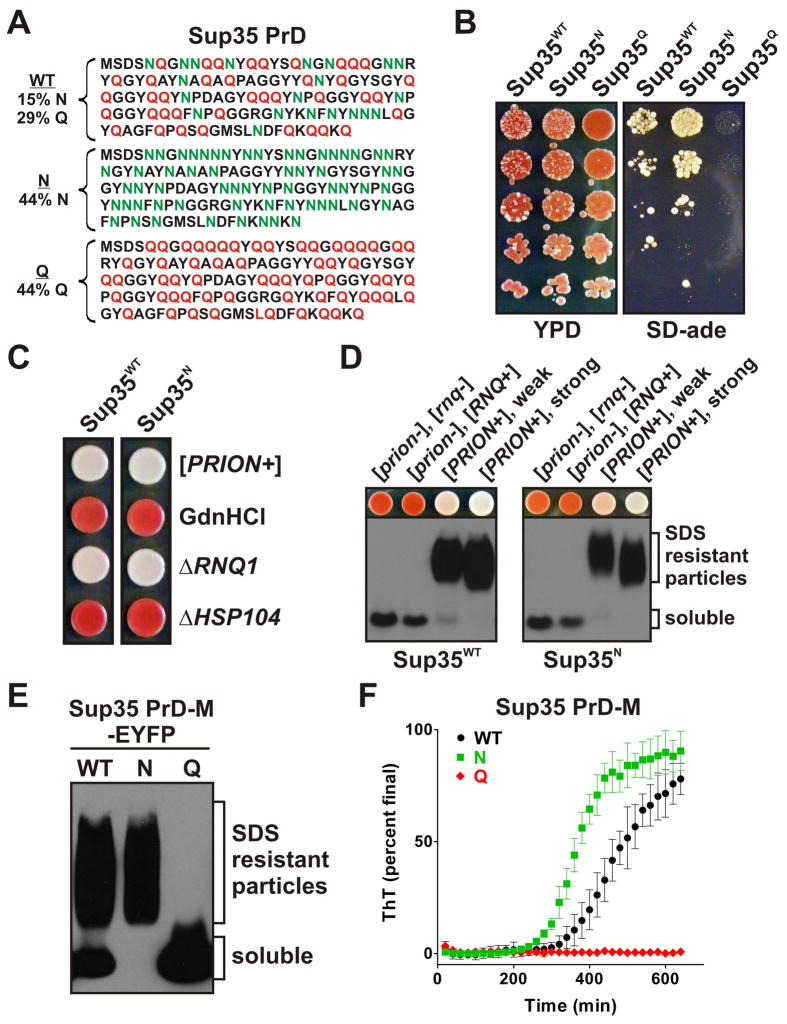
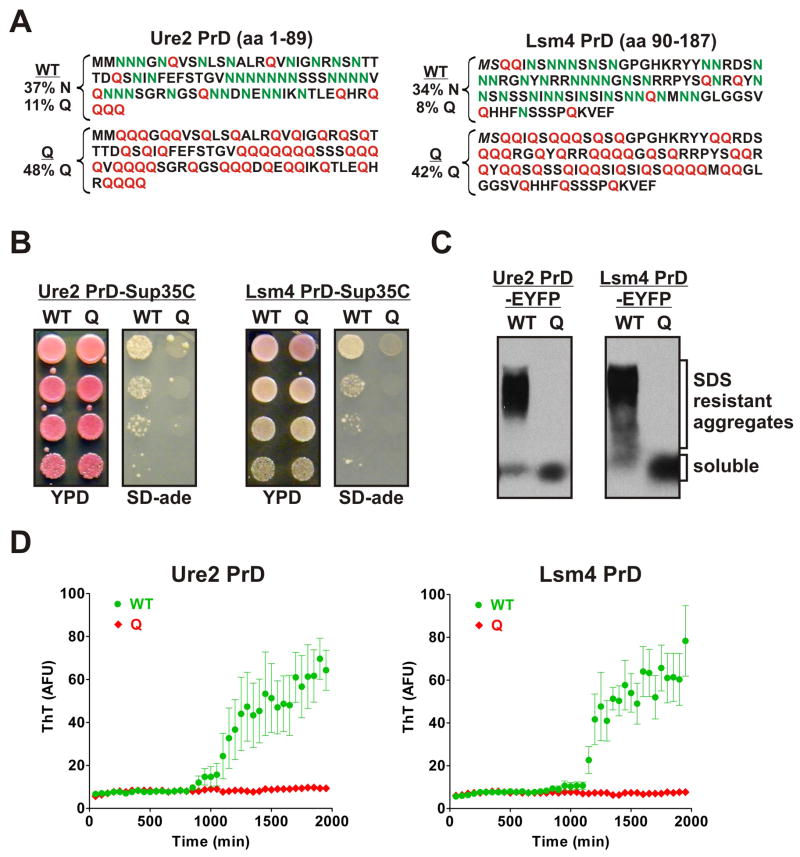
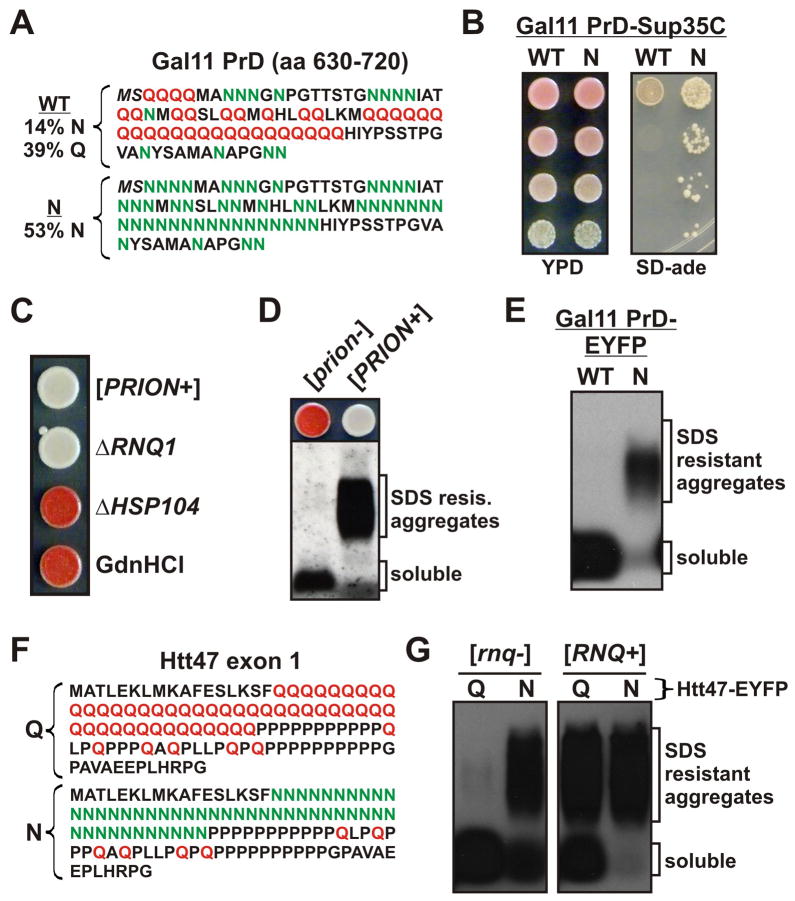
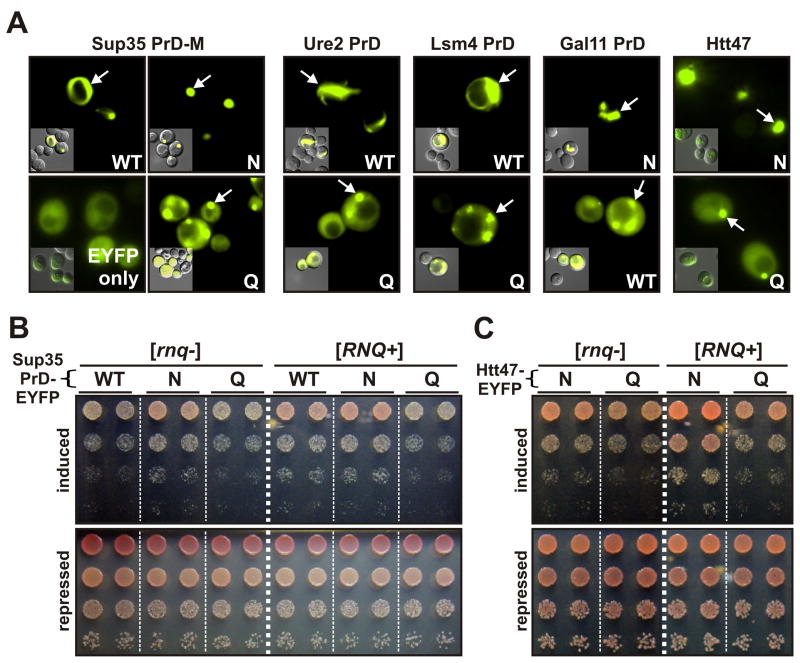
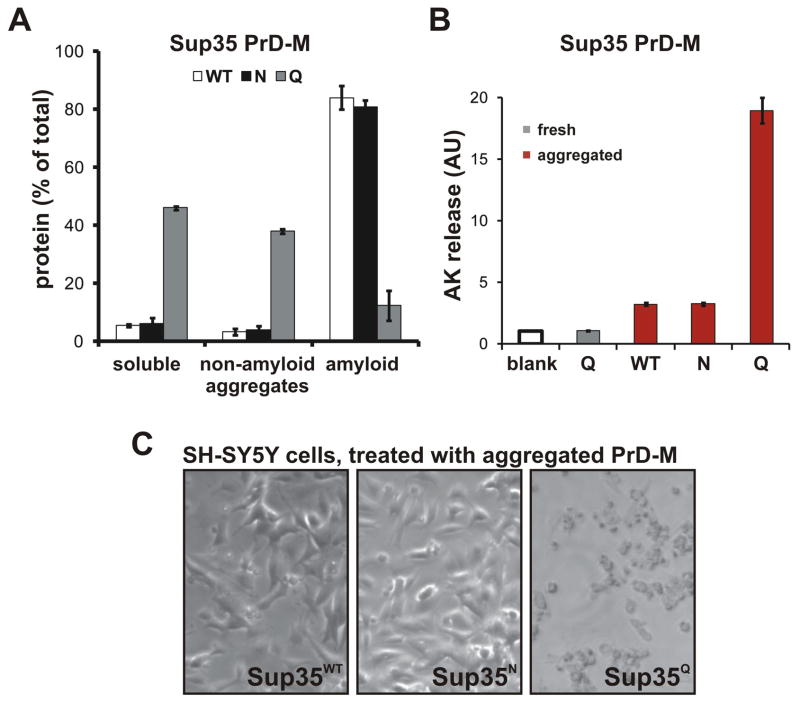
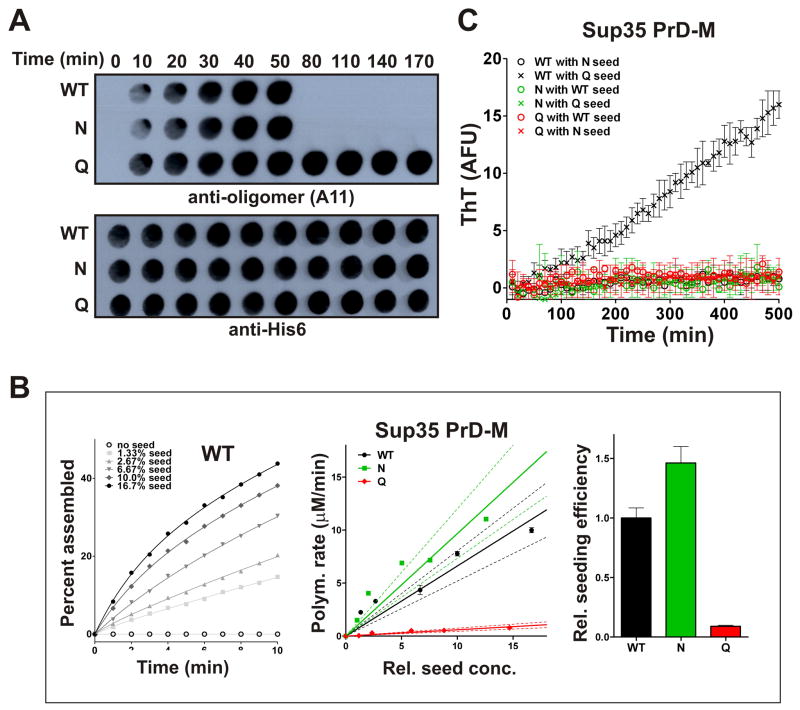
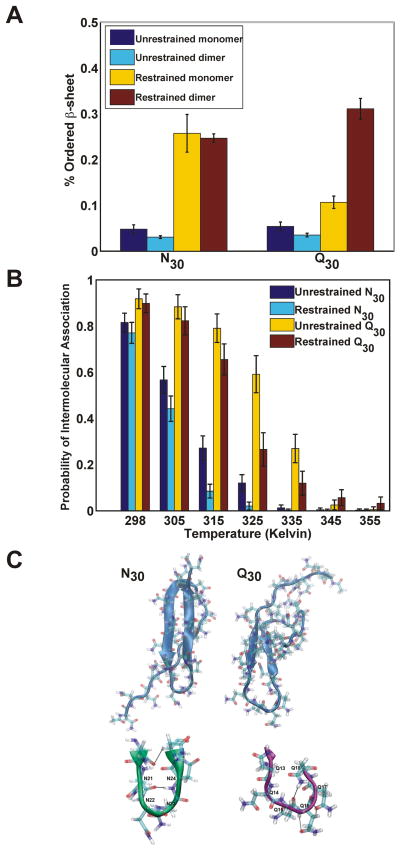
Sequences rich in glutamine (Q) and asparagine (N) residues often fail to fold at the monomer level. This, coupled to their unusual hydrogen-bonding abilities, provides the driving force to switch between disordered monomers and amyloids. Such transitions govern processes as diverse as human protein-folding diseases, bacterial biofilm assembly, and the inheritance of yeast prions (protein-based genetic elements). A systematic survey of prion-forming domains suggested that Q and N residues have distinct effects on amyloid formation. Here, we use cell biological, biochemical, and computational techniques to compare Q/N-rich protein variants, replacing Ns with Qs and Qs with Ns. We find that the two residues have strong and opposing effects: N richness promotes assembly of benign self-templating amyloids; Q richness promotes formation of toxic nonamyloid conformers. Molecular simulations focusing on intrinsic folding differences between Qs and Ns suggest that their different behaviors are due to the enhanced turn-forming propensity of Ns over Qs.
Copyright © 2011 Elsevier Inc. All rights reserved.
Figures
References
-
- Allen KD, Chernova TA, Tennant EP, Wilkinson KD, Chernoff YO. Effects of ubiquitin system alterations on the formation and loss of a yeast prion. J Biol Chem. 2007;282:3004–3013. - PubMed
-
- Chernoff YO, Lindquist SL, Ono B, Inge-Vechtomov SG, Liebman SW. Role of the chaperone protein Hsp104 in propagation of the yeast prion-like factor [psi+] Science. 1995;268:880–884. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
