

ASF1A and ATM regulate H3K56-mediated cell-cycle checkpoint recovery in response to UV irradiation
- PMID: 21727091
- PMCID: PMC3185425
- DOI: 10.1093/nar/gkr523
ASF1A and ATM regulate H3K56-mediated cell-cycle checkpoint recovery in response to UV irradiation
Abstract
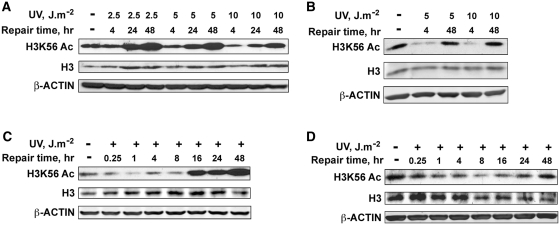
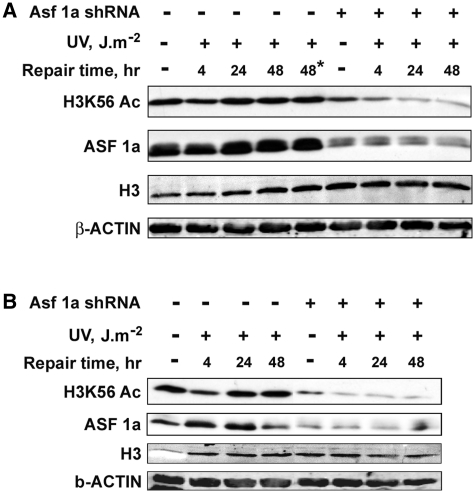
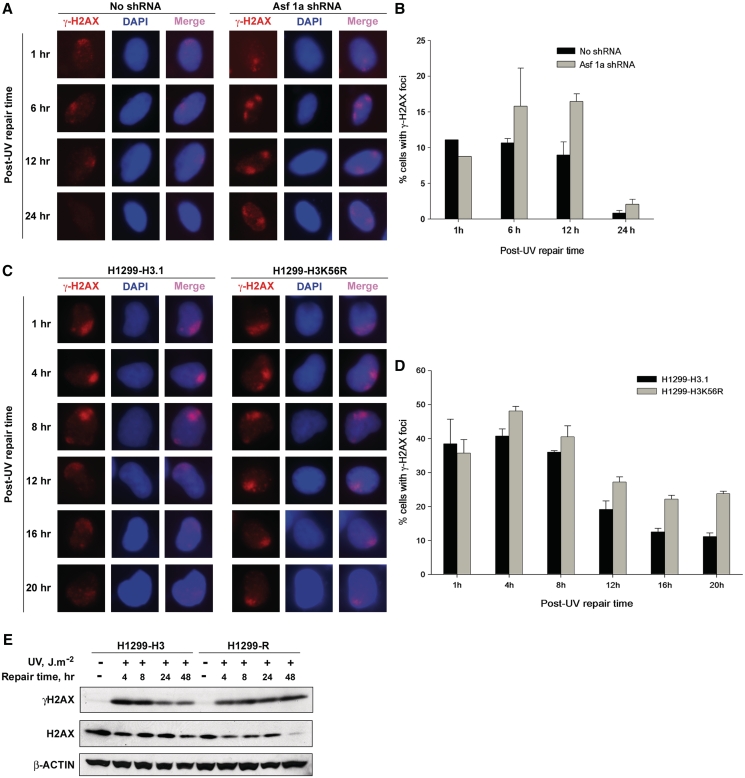
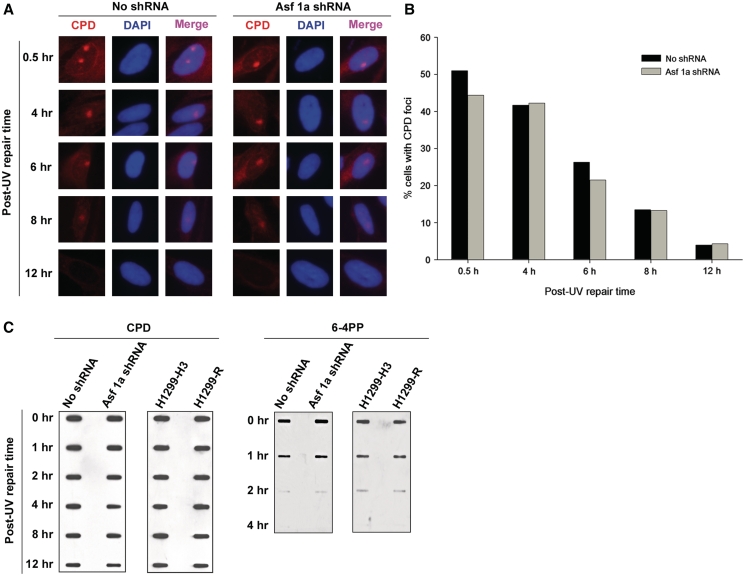
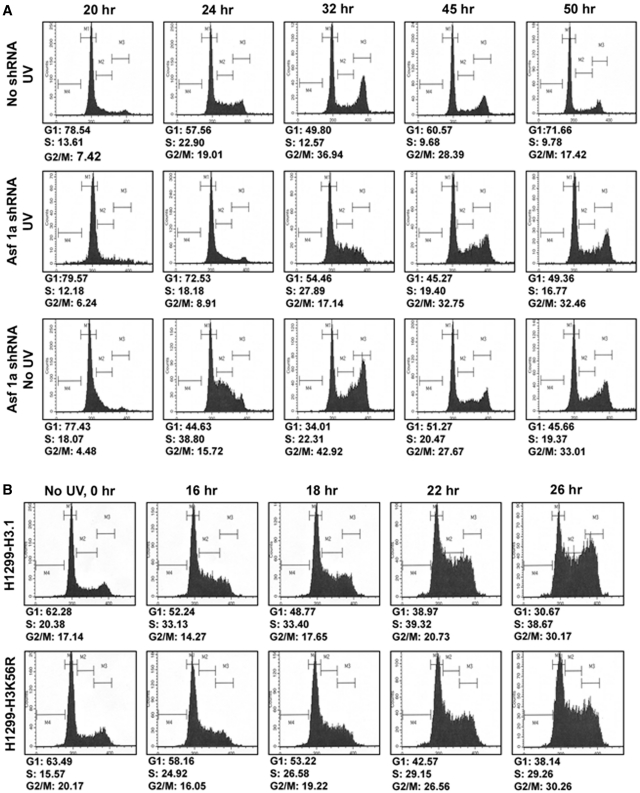
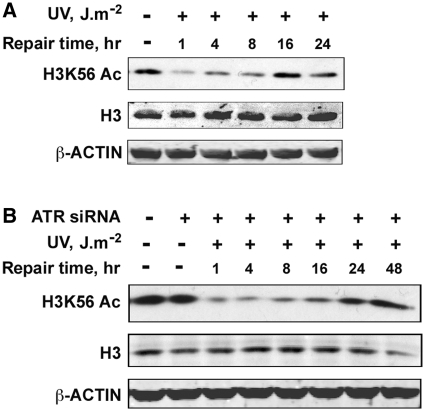
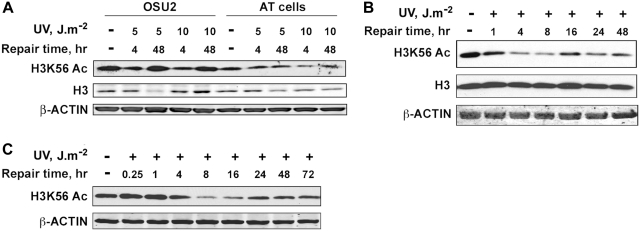
Successful DNA repair within chromatin requires coordinated interplay of histone modifications, chaperones and remodelers for allowing access of repair and checkpoint machineries to damaged sites. Upon completion of repair, ordered restoration of chromatin structure and key epigenetic marks herald the cell's normal function. Here, we demonstrate such a restoration role of H3K56 acetylation (H3K56Ac) mark in response to ultraviolet (UV) irradiation of human cells. A fast initial deacetylation of H3K56 is followed by full renewal of an acetylated state at ~24-48 h post-irradiation. Histone chaperone, anti-silencing function-1 A (ASF1A), is crucial for post-repair H3K56Ac restoration, which in turn, is needed for the dephosphorylation of γ-H2AX and cellular recovery from checkpoint arrest. On the other hand, completion of DNA damage repair is not dependent on ASF1A or H3K56Ac. H3K56Ac restoration is regulated by ataxia telangiectasia mutated (ATM) checkpoint kinase. These cross-talking molecular cellular events reveal the important pathway components influencing the regulatory function of H3K56Ac in the recovery from UV-induced checkpoint arrest.
Figures

References
-
- Harper JW, Elledge SJ. The DNA damage response: ten years after. Mol. Cell. 2007;28:739–745. - PubMed
-
- De Boer J, Hoeijmakers JH. Nucleotide excision repair and human syndromes. car. 2000;21:453–460. - PubMed
-
- Reardon JT, Sancar A. Nucleotide excision repair. Prog. Nucleic Acid Res. Mol. Biol. 2005;79:183–235. - PubMed
-
- Mitchell DL, Haipek CA, Clarkson JM. Photoproducts are removed from the DNA of UV-irradiated mammalian cells more efficiently than cyclobutane pyrimidine dimers. Mutat. Res. 1985;143:109–112. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
