

Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis
- PMID: 21727188
- PMCID: PMC3149226
- DOI: 10.1084/jem.20110958
Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis
Abstract
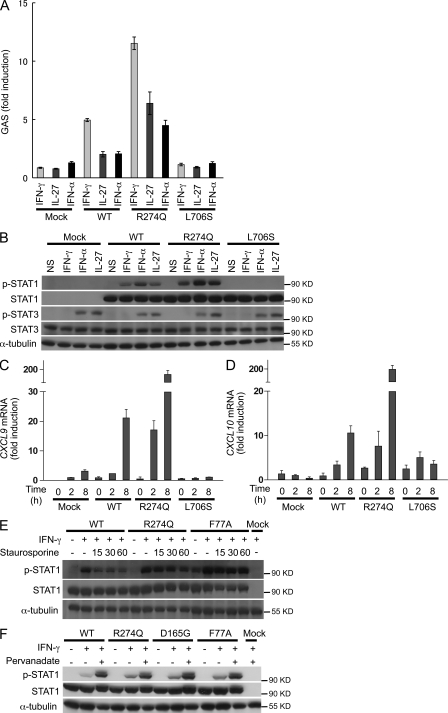
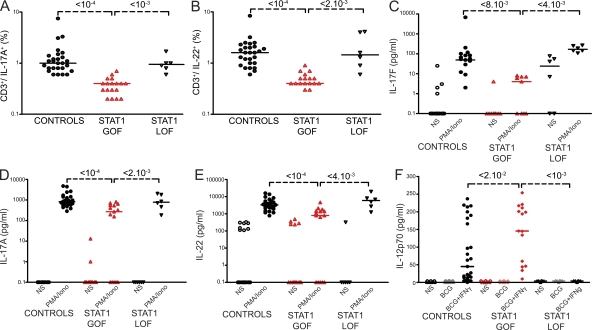
Chronic mucocutaneous candidiasis disease (CMCD) may be caused by autosomal dominant (AD) IL-17F deficiency or autosomal recessive (AR) IL-17RA deficiency. Here, using whole-exome sequencing, we identified heterozygous germline mutations in STAT1 in 47 patients from 20 kindreds with AD CMCD. Previously described heterozygous STAT1 mutant alleles are loss-of-function and cause AD predisposition to mycobacterial disease caused by impaired STAT1-dependent cellular responses to IFN-γ. Other loss-of-function STAT1 alleles cause AR predisposition to intracellular bacterial and viral diseases, caused by impaired STAT1-dependent responses to IFN-α/β, IFN-γ, IFN-λ, and IL-27. In contrast, the 12 AD CMCD-inducing STAT1 mutant alleles described here are gain-of-function and increase STAT1-dependent cellular responses to these cytokines, and to cytokines that predominantly activate STAT3, such as IL-6 and IL-21. All of these mutations affect the coiled-coil domain and impair the nuclear dephosphorylation of activated STAT1, accounting for their gain-of-function and dominance. Stronger cellular responses to the STAT1-dependent IL-17 inhibitors IFN-α/β, IFN-γ, and IL-27, and stronger STAT1 activation in response to the STAT3-dependent IL-17 inducers IL-6 and IL-21, hinder the development of T cells producing IL-17A, IL-17F, and IL-22. Gain-of-function STAT1 alleles therefore cause AD CMCD by impairing IL-17 immunity.
Figures


References
-
- Atkinson T.P., Schäffer A.A., Grimbacher B., Schroeder H.W., Jr, Woellner C., Zerbe C.S., Puck J.M. 2001. An immune defect causing dominant chronic mucocutaneous candidiasis and thyroid disease maps to chromosome 2p in a single family. Am. J. Hum. Genet. 69:791–803 10.1086/323611 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
