

Comparative analysis of methods for detecting interacting loci
- PMID: 21729295
- PMCID: PMC3161015
- DOI: 10.1186/1471-2164-12-344
Comparative analysis of methods for detecting interacting loci
Abstract
Background: Interactions among genetic loci are believed to play an important role in disease risk. While many methods have been proposed for detecting such interactions, their relative performance remains largely unclear, mainly because different data sources, detection performance criteria, and experimental protocols were used in the papers introducing these methods and in subsequent studies. Moreover, there have been very few studies strictly focused on comparison of existing methods. Given the importance of detecting gene-gene and gene-environment interactions, a rigorous, comprehensive comparison of performance and limitations of available interaction detection methods is warranted.
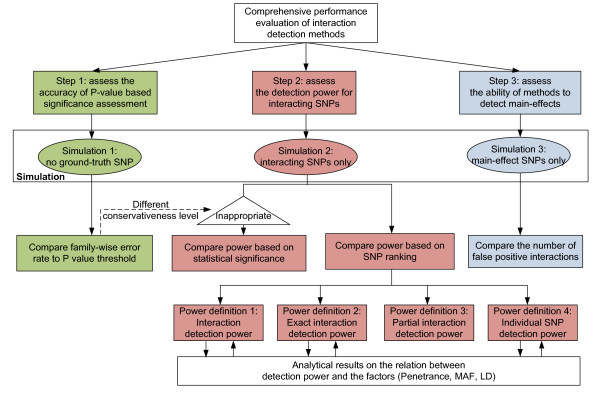
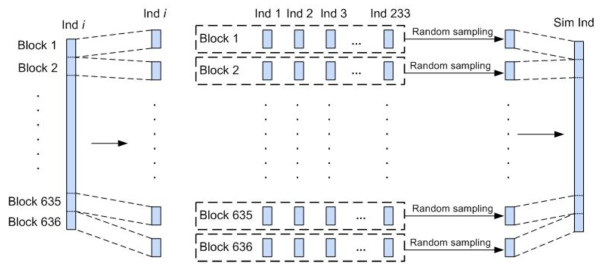
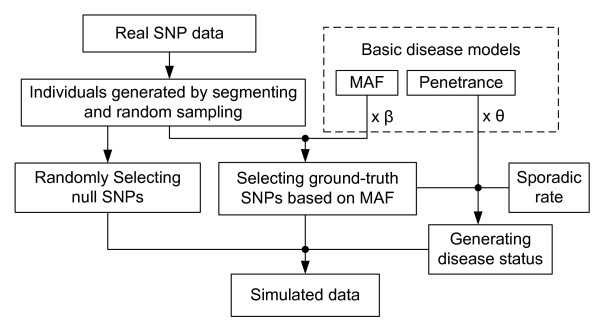
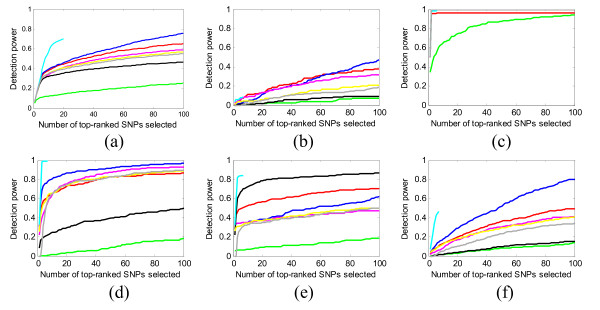
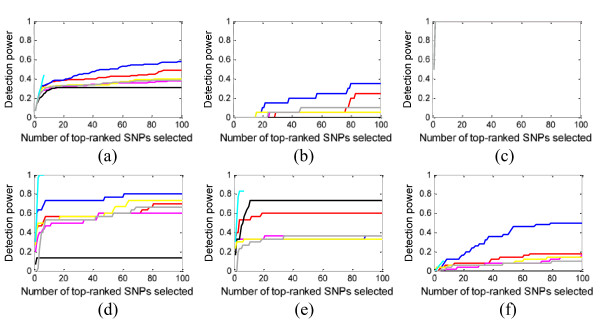
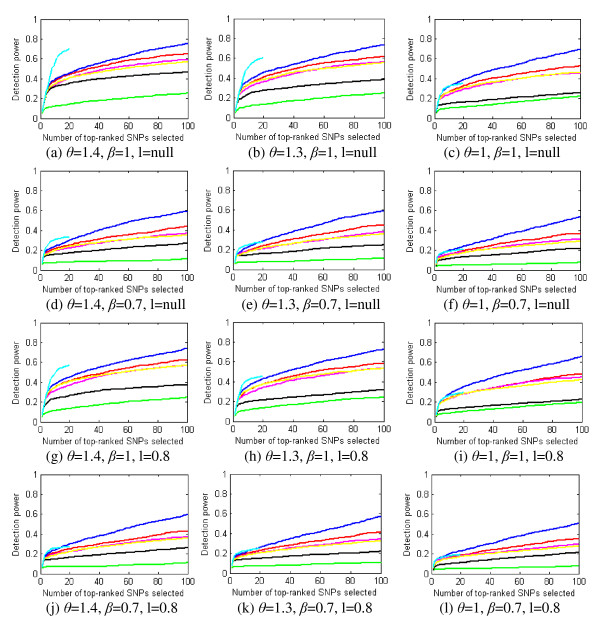
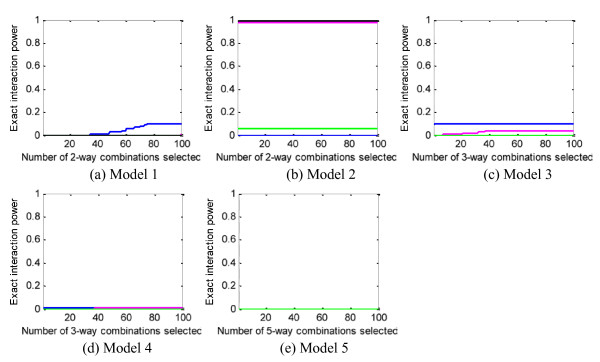
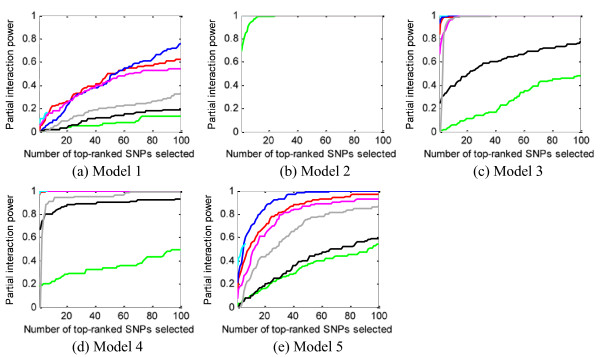
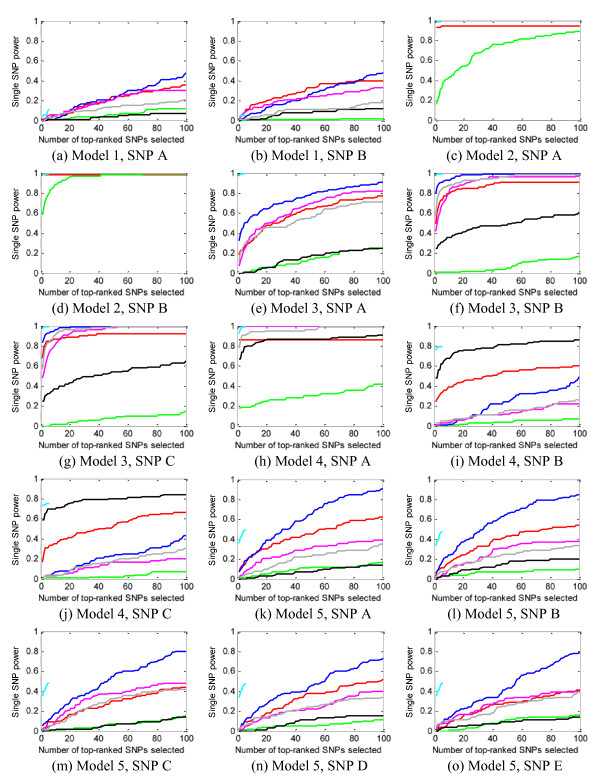
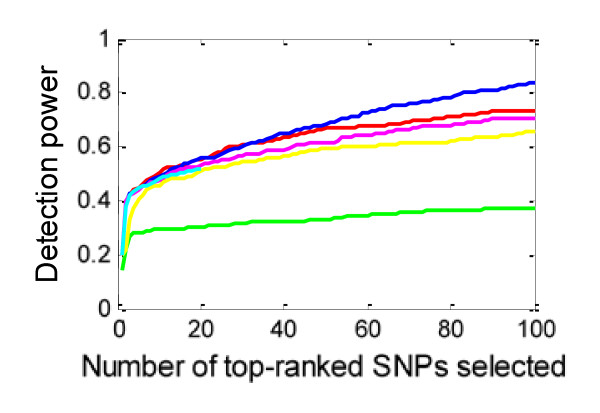
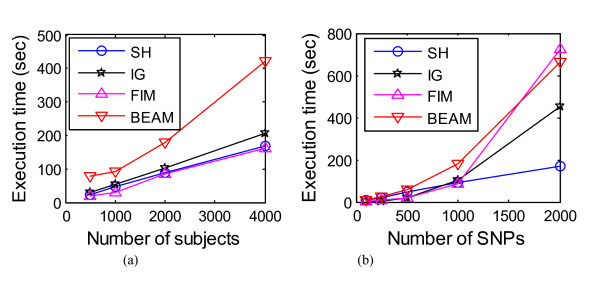
Results: We report a comparison of eight representative methods, of which seven were specifically designed to detect interactions among single nucleotide polymorphisms (SNPs), with the last a popular main-effect testing method used as a baseline for performance evaluation. The selected methods, multifactor dimensionality reduction (MDR), full interaction model (FIM), information gain (IG), Bayesian epistasis association mapping (BEAM), SNP harvester (SH), maximum entropy conditional probability modeling (MECPM), logistic regression with an interaction term (LRIT), and logistic regression (LR) were compared on a large number of simulated data sets, each, consistent with complex disease models, embedding multiple sets of interacting SNPs, under different interaction models. The assessment criteria included several relevant detection power measures, family-wise type I error rate, and computational complexity. There are several important results from this study. First, while some SNPs in interactions with strong effects are successfully detected, most of the methods miss many interacting SNPs at an acceptable rate of false positives. In this study, the best-performing method was MECPM. Second, the statistical significance assessment criteria, used by some of the methods to control the type I error rate, are quite conservative, thereby limiting their power and making it difficult to fairly compare them. Third, as expected, power varies for different models and as a function of penetrance, minor allele frequency, linkage disequilibrium and marginal effects. Fourth, the analytical relationships between power and these factors are derived, aiding in the interpretation of the study results. Fifth, for these methods the magnitude of the main effect influences the power of the tests. Sixth, most methods can detect some ground-truth SNPs but have modest power to detect the whole set of interacting SNPs.
Conclusion: This comparison study provides new insights into the strengths and limitations of current methods for detecting interacting loci. This study, along with freely available simulation tools we provide, should help support development of improved methods. The simulation tools are available at: http://code.google.com/p/simulation-tool-bmc-ms9169818735220977/downloads/list.
Figures
References
-
- Hirschhorn J. Genome-wide association studies for common diseases and complex traits. Nature reviews Genetics. 2005;6:95–108. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
