

A new way to see RNA
- PMID: 21729350
- PMCID: PMC4410278
- DOI: 10.1017/S0033583511000059
A new way to see RNA
Abstract
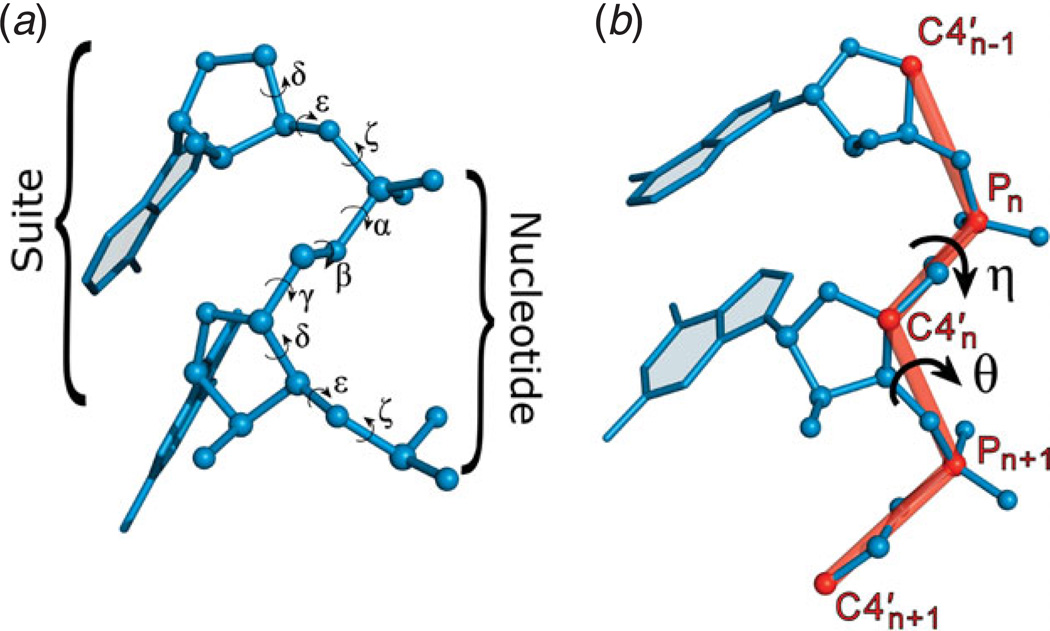
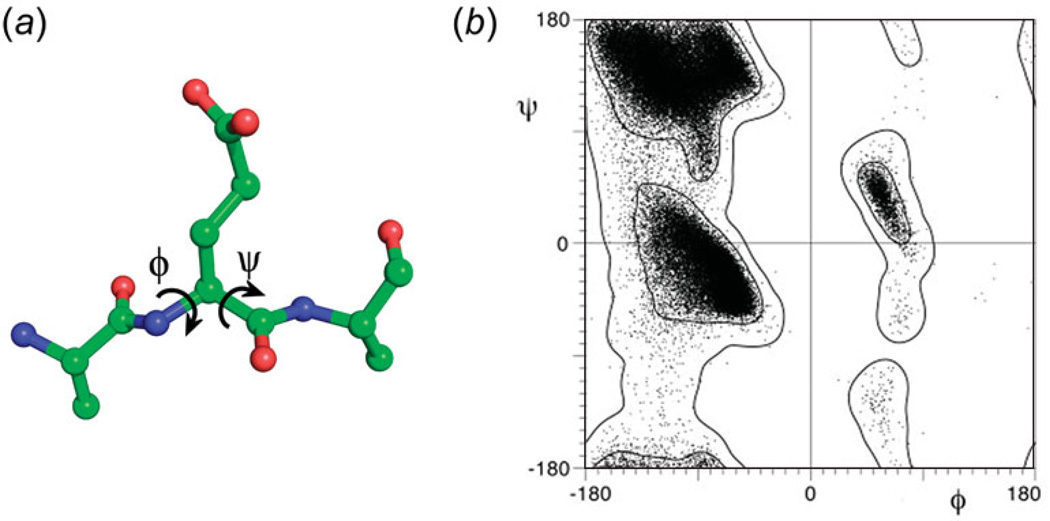
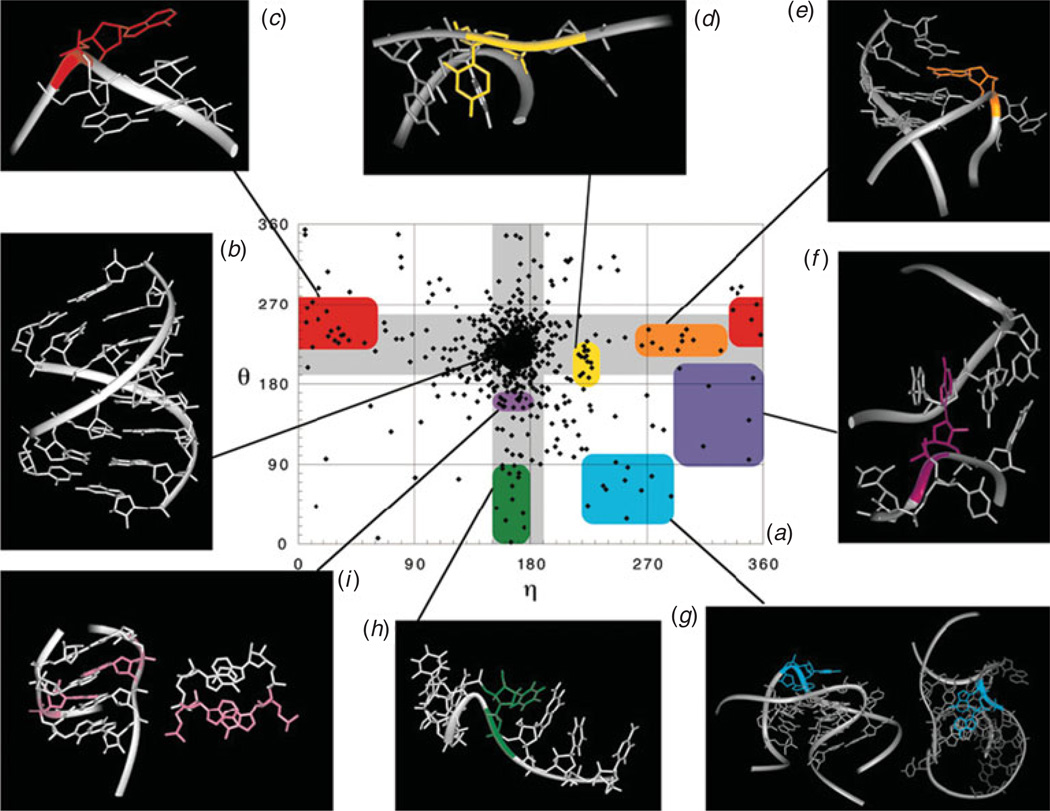
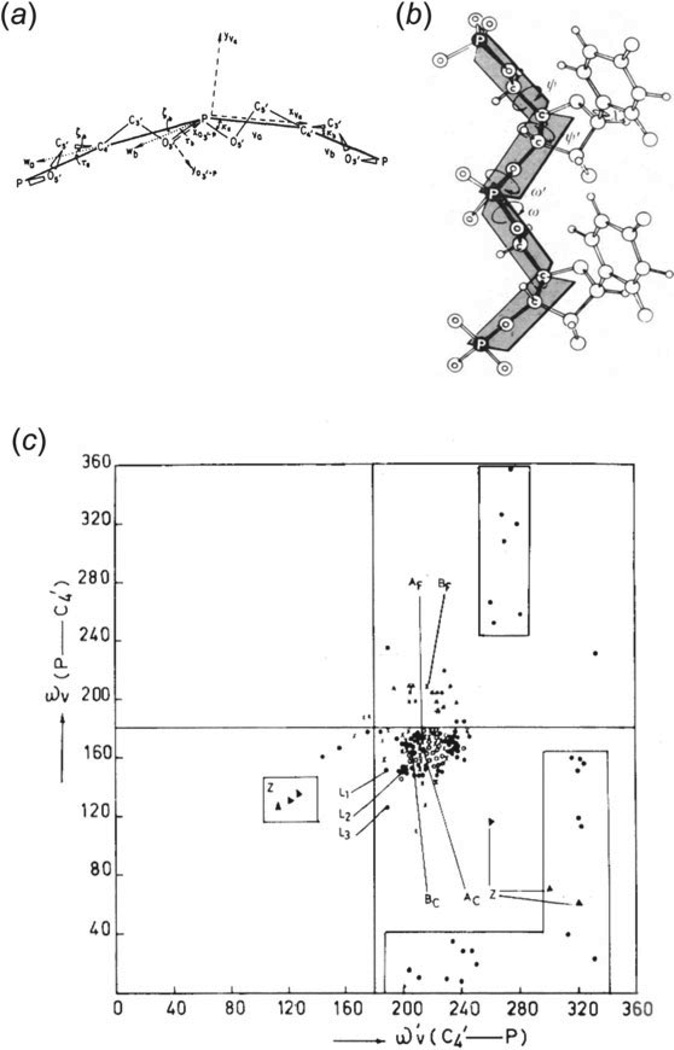
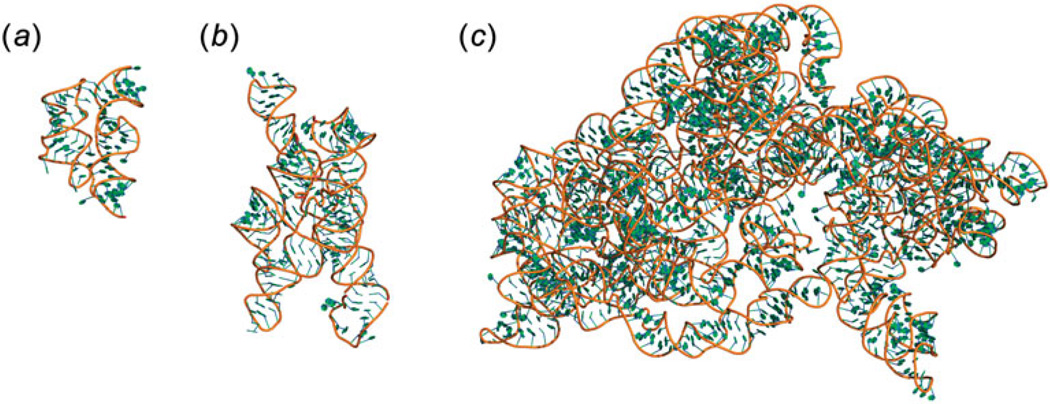
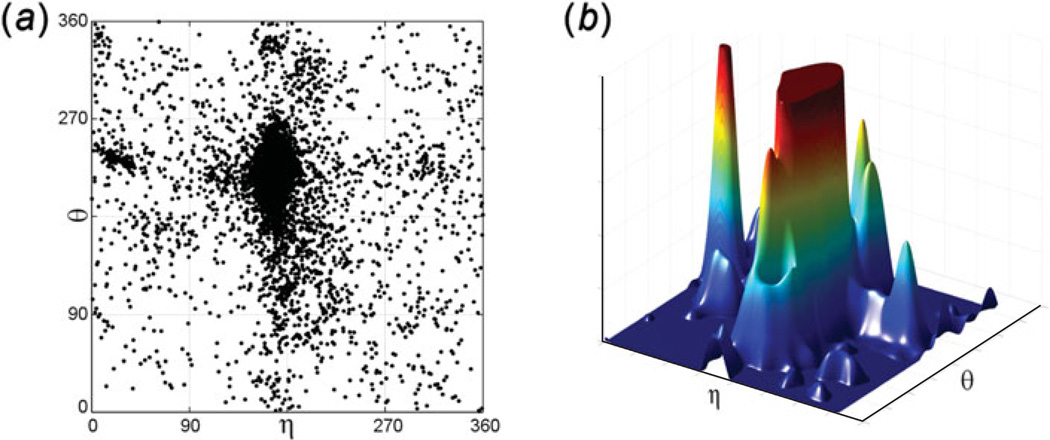
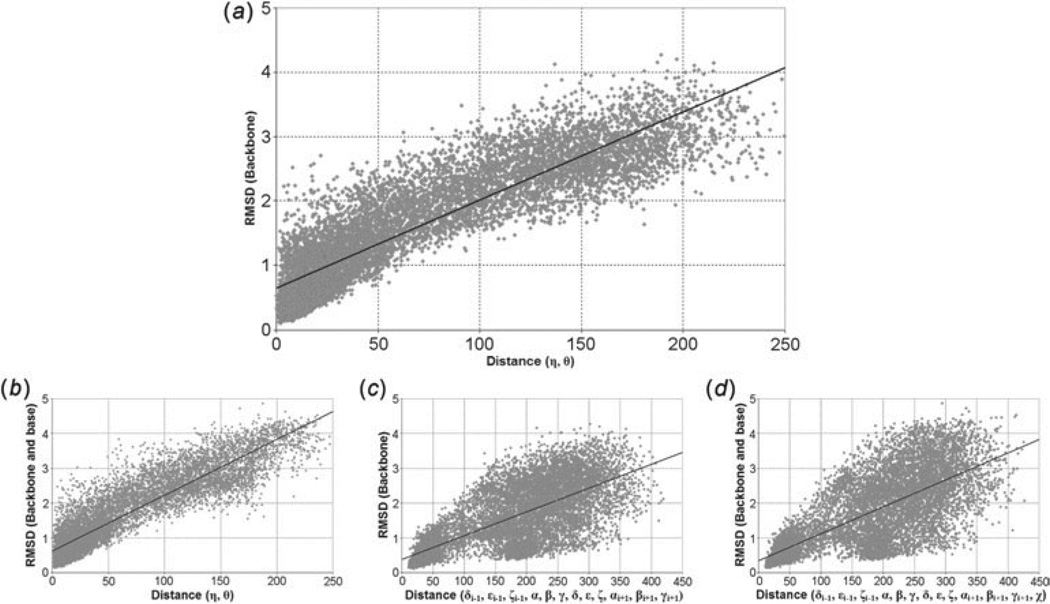
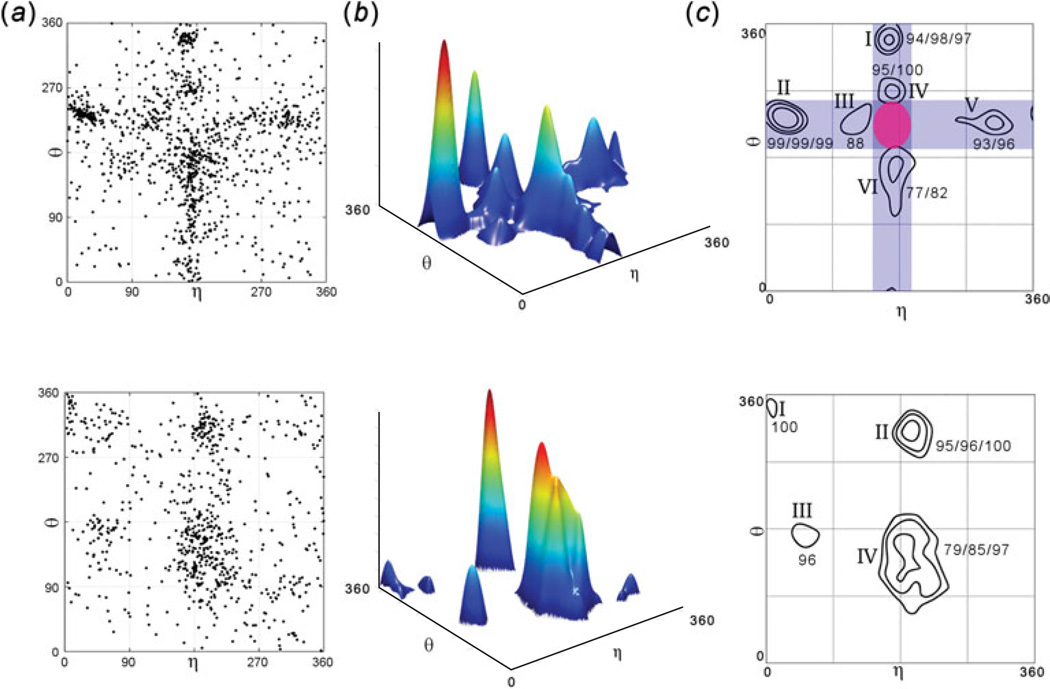
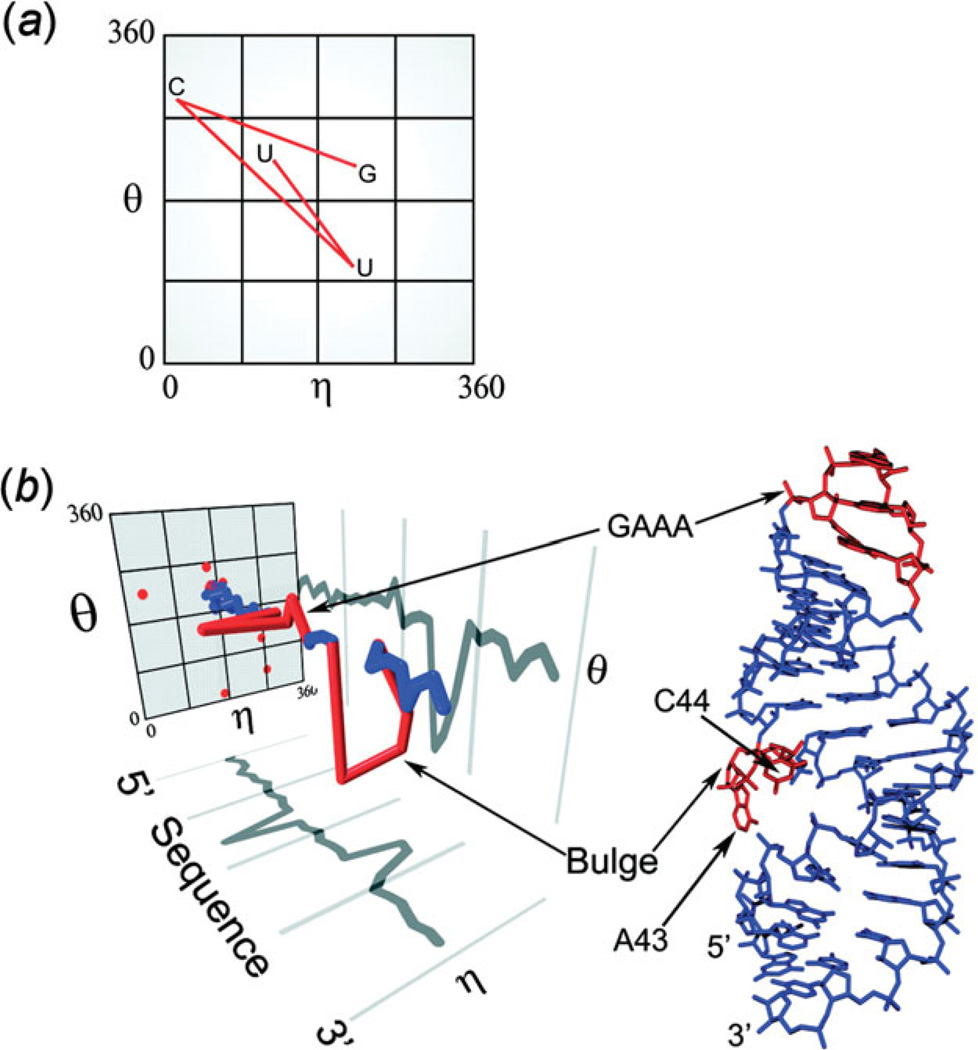
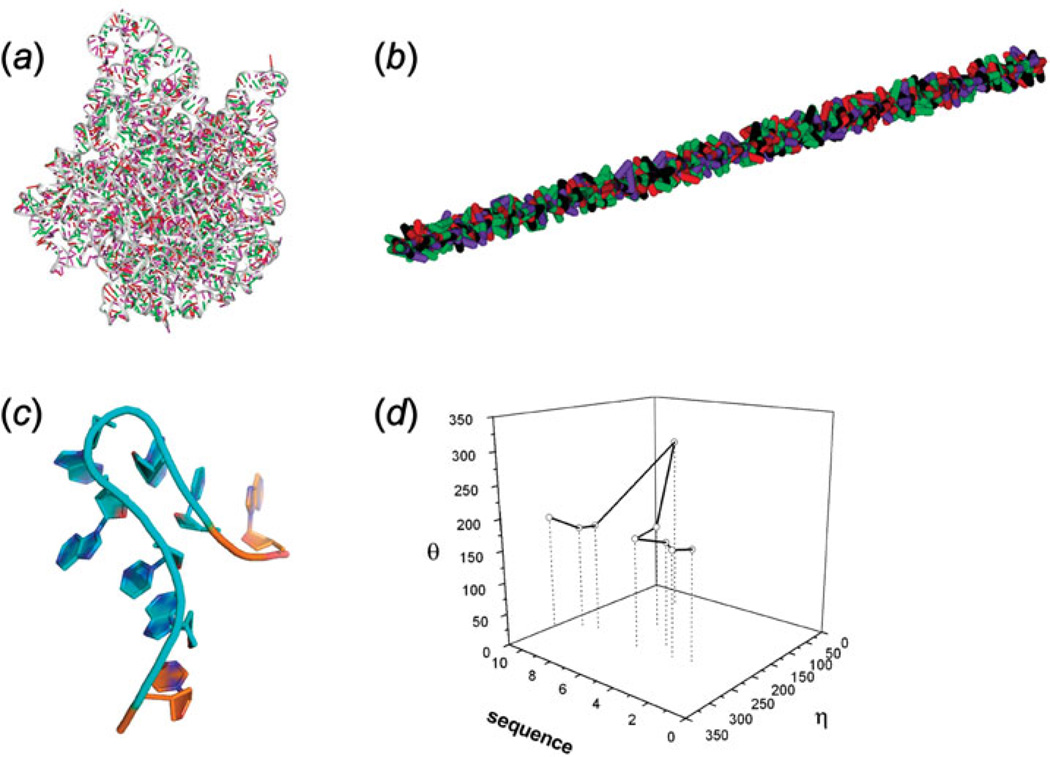
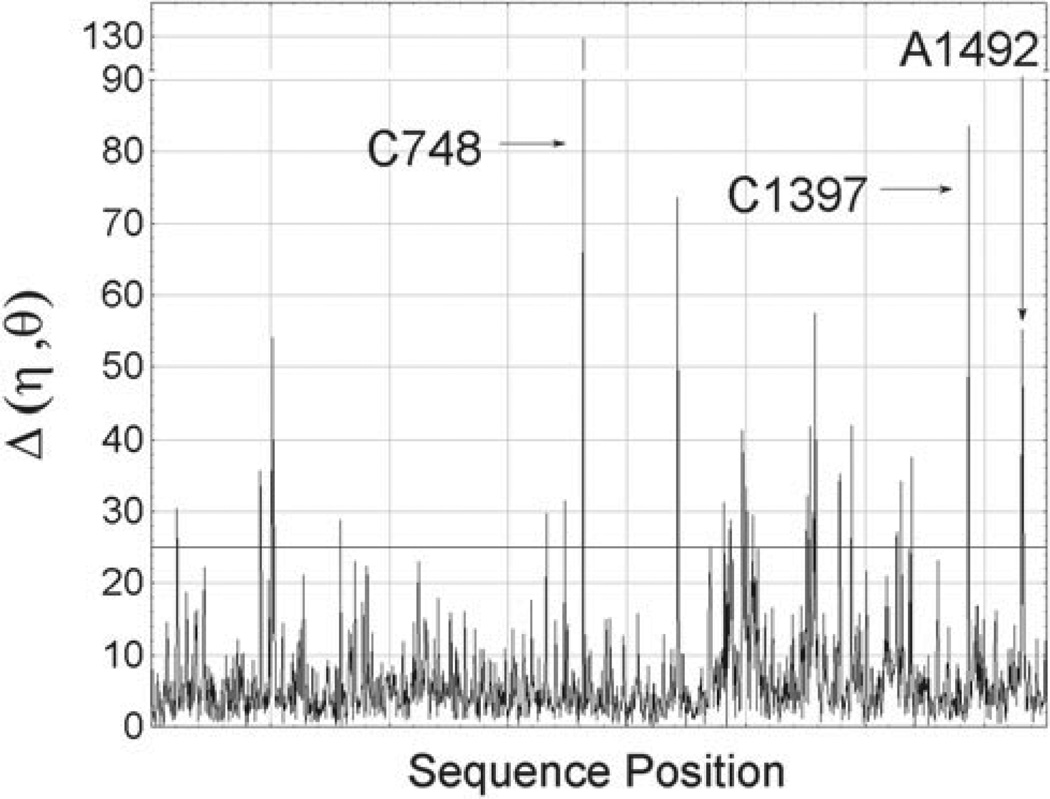
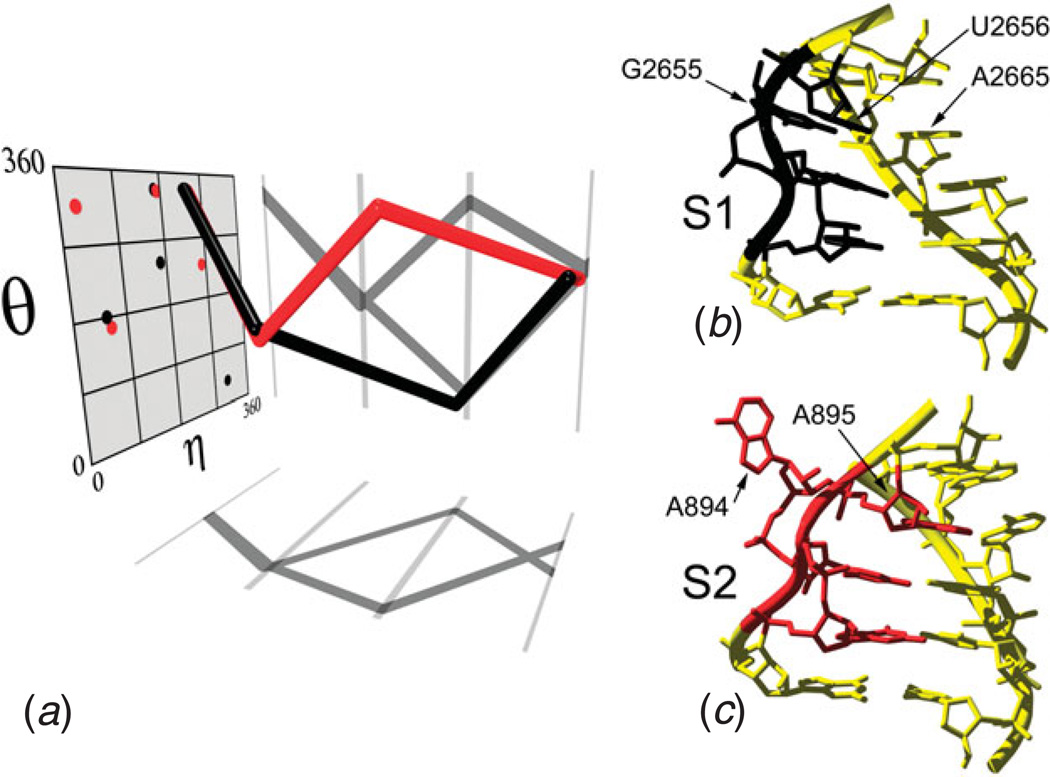
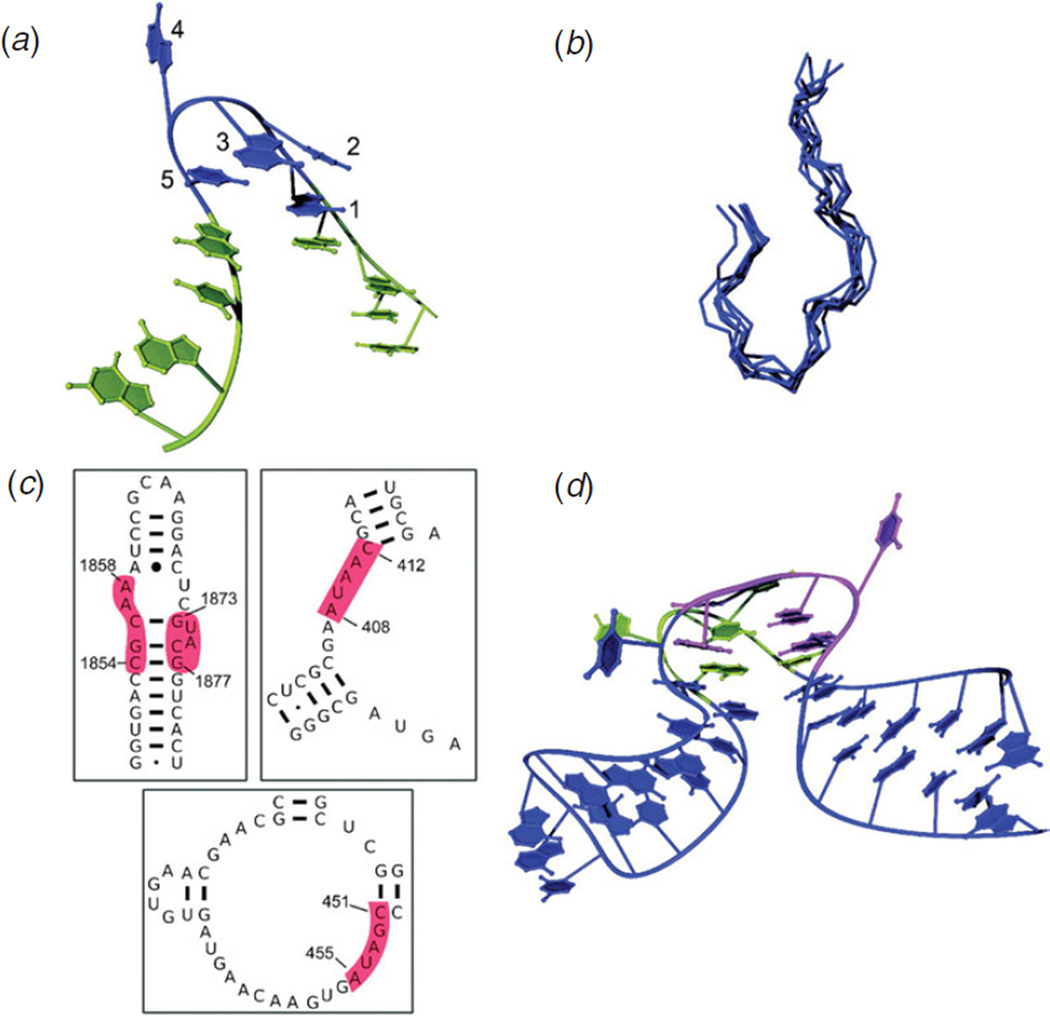
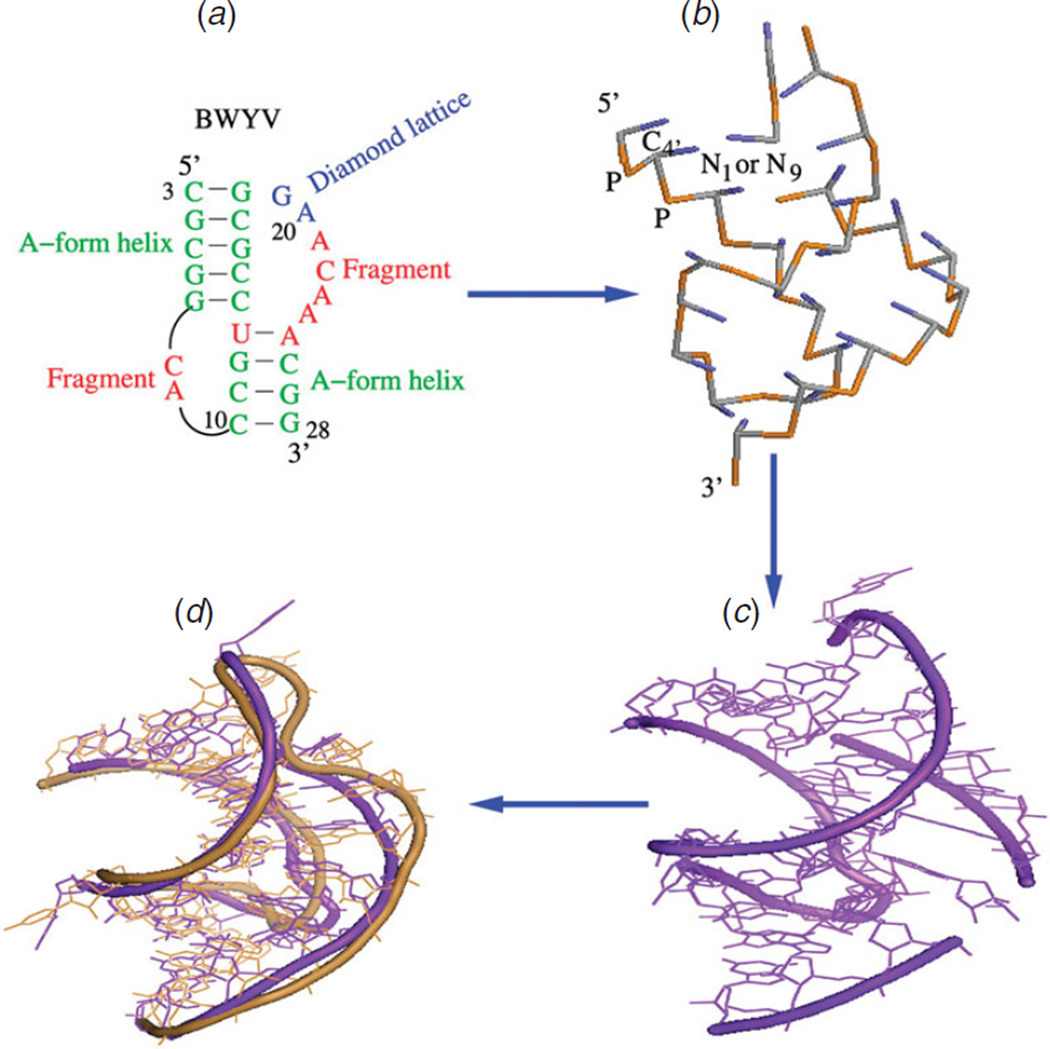
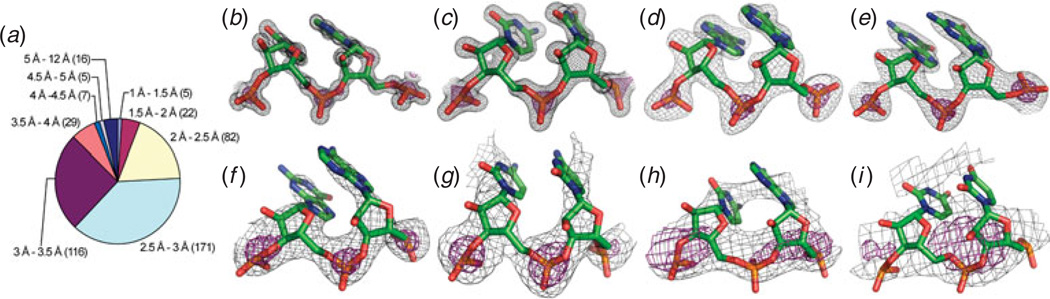
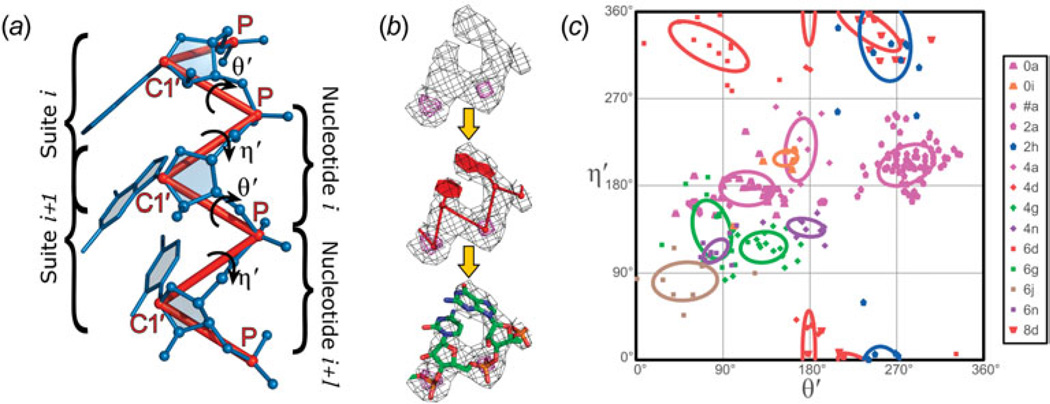
Unlike proteins, the RNA backbone has numerous degrees of freedom (eight, if one counts the sugar pucker), making RNA modeling, structure building and prediction a multidimensional problem of exceptionally high complexity. And yet RNA tertiary structures are not infinite in their structural morphology; rather, they are built from a limited set of discrete units. In order to reduce the dimensionality of the RNA backbone in a physically reasonable way, a shorthand notation was created that reduced the RNA backbone torsion angles to two (η and θ, analogous to φ and ψ in proteins). When these torsion angles are calculated for nucleotides in a crystallographic database and plotted against one another, one obtains a plot analogous to a Ramachandran plot (the η/θ plot), with highly populated and unpopulated regions. Nucleotides that occupy proximal positions on the plot have identical structures and are found in the same units of tertiary structure. In this review, we describe the statistical validation of the η/θ formalism and the exploration of features within the η/θ plot. We also describe the application of the η/θ formalism in RNA motif discovery, structural comparison, RNA structure building and tertiary structure prediction. More than a tool, however, the η/θ formalism has provided new insights into RNA structure itself, revealing its fundamental components and the factors underlying RNA architectural form.
Figures
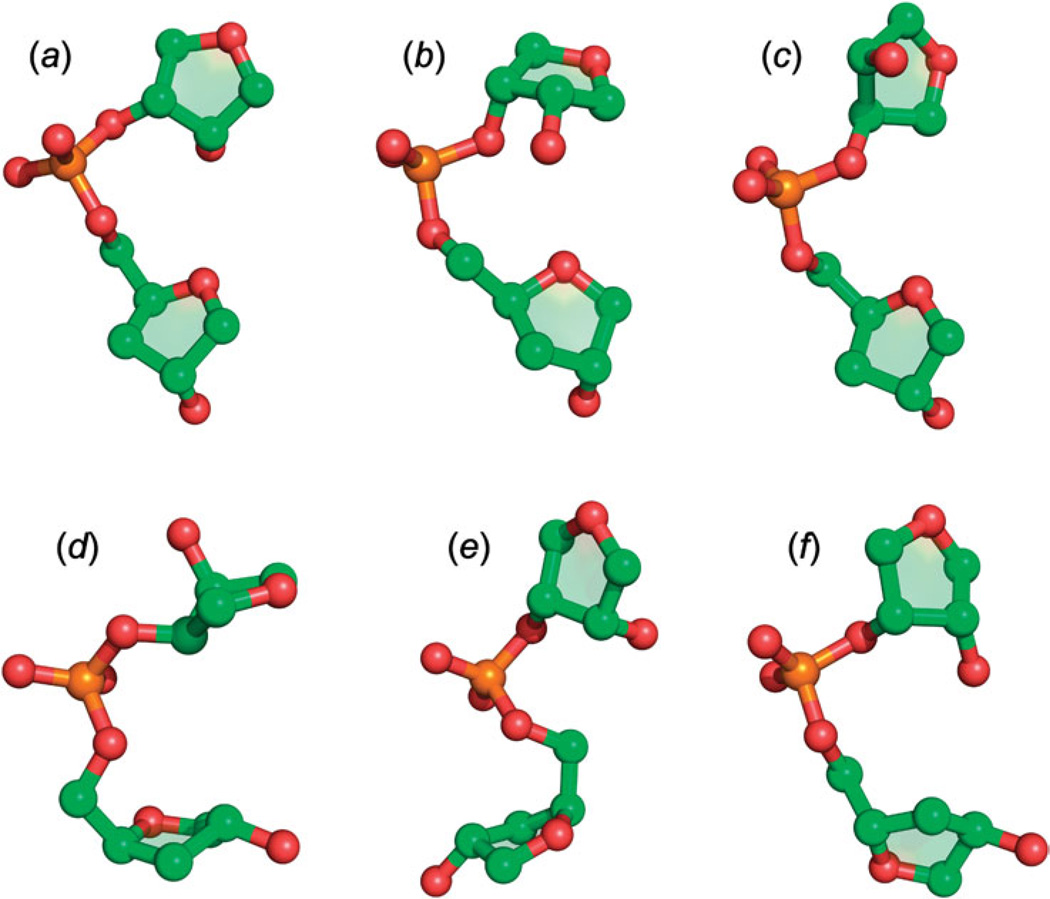
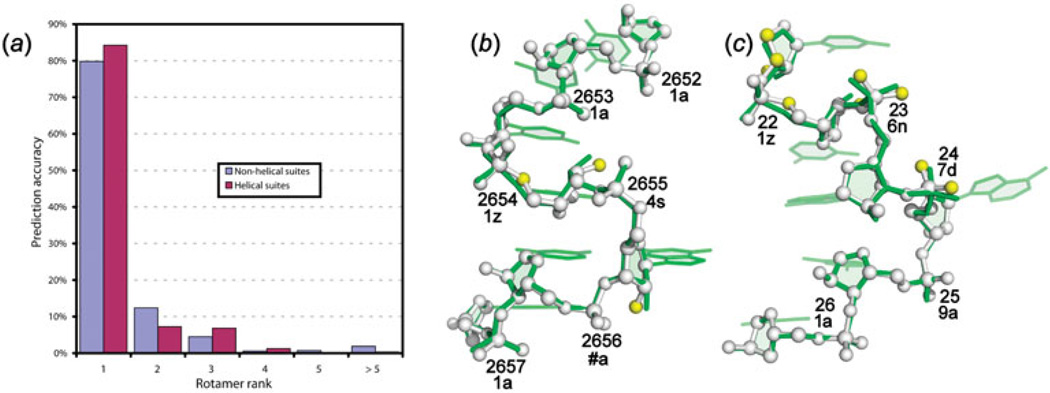
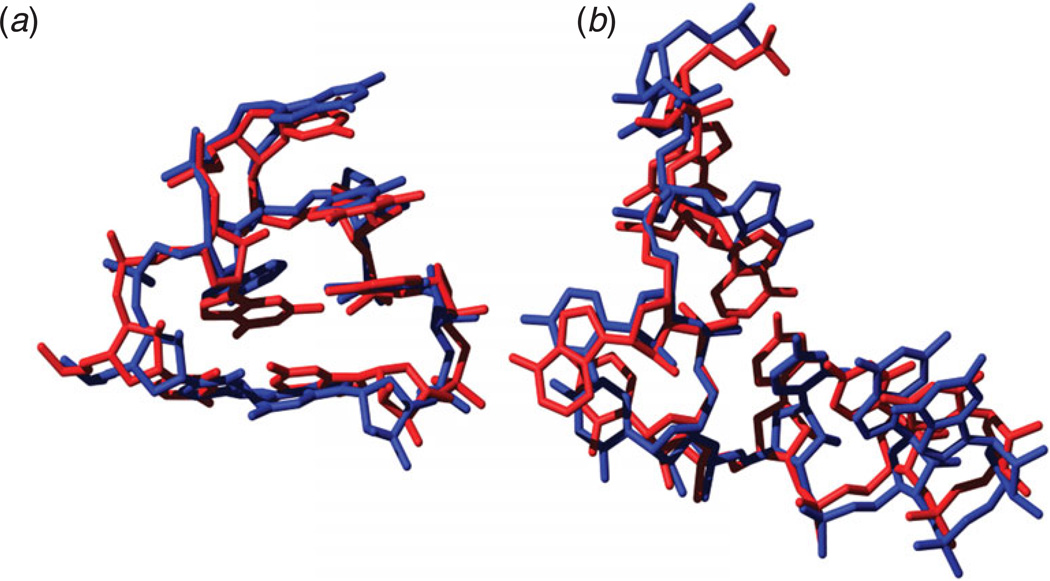
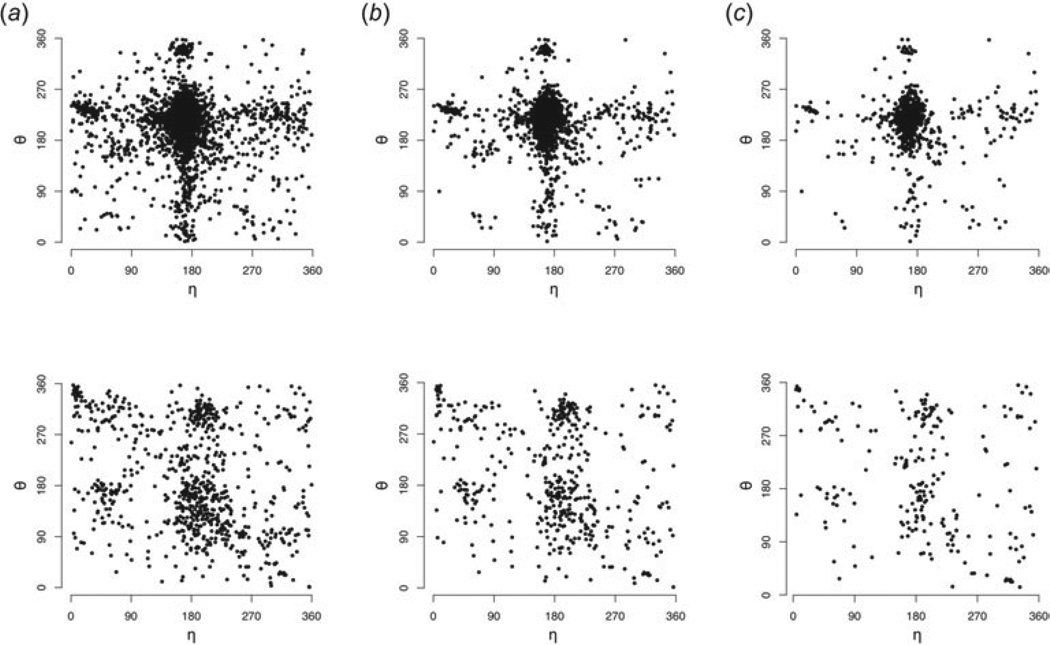
Similar articles
-
Evaluating and learning from RNA pseudotorsional space: quantitative validation of a reduced representation for RNA structure.J Mol Biol. 2007 Sep 28;372(4):942-957. doi: 10.1016/j.jmb.2007.06.058. Epub 2007 Jun 27. J Mol Biol. 2007. PMID: 17707400 Free PMC article.
-
Stepping through an RNA structure: A novel approach to conformational analysis.J Mol Biol. 1998 Dec 18;284(5):1465-78. doi: 10.1006/jmbi.1998.2233. J Mol Biol. 1998. PMID: 9878364
-
AMIGOS III: pseudo-torsion angle visualization and motif-based structure comparison of nucleic acids.Bioinformatics. 2022 May 13;38(10):2937-2939. doi: 10.1093/bioinformatics/btac207. Bioinformatics. 2022. PMID: 35561202 Free PMC article.
-
The RNA 3D Motif Atlas: Computational methods for extraction, organization and evaluation of RNA motifs.Methods. 2016 Jul 1;103:99-119. doi: 10.1016/j.ymeth.2016.04.025. Epub 2016 Apr 25. Methods. 2016. PMID: 27125735 Free PMC article. Review.
-
Modeling the three-dimensional structure of RNA.FASEB J. 1993 Jan;7(1):97-105. doi: 10.1096/fasebj.7.1.7678567. FASEB J. 1993. PMID: 7678567 Review.
Cited by
-
Mapping L1 ligase ribozyme conformational switch.J Mol Biol. 2012 Oct 12;423(1):106-22. doi: 10.1016/j.jmb.2012.06.035. Epub 2012 Jul 3. J Mol Biol. 2012. PMID: 22771572 Free PMC article.
-
Understanding the binding specificities of mRNA targets by the mammalian Quaking protein.Nucleic Acids Res. 2019 Nov 18;47(20):10564-10579. doi: 10.1093/nar/gkz877. Nucleic Acids Res. 2019. PMID: 31602485 Free PMC article.
-
Web 3DNA 2.0 for the analysis, visualization, and modeling of 3D nucleic acid structures.Nucleic Acids Res. 2019 Jul 2;47(W1):W26-W34. doi: 10.1093/nar/gkz394. Nucleic Acids Res. 2019. PMID: 31114927 Free PMC article.
-
Atomic structure of potato virus X, the prototype of the Alphaflexiviridae family.Nat Chem Biol. 2020 May;16(5):564-569. doi: 10.1038/s41589-020-0502-4. Epub 2020 Mar 16. Nat Chem Biol. 2020. PMID: 32203412
-
Origins of Life: The Protein Folding Problem all over again?Proc Natl Acad Sci U S A. 2024 Aug 20;121(34):e2315000121. doi: 10.1073/pnas.2315000121. Epub 2024 Aug 12. Proc Natl Acad Sci U S A. 2024. PMID: 39133848 Free PMC article.
References
-
- Abramovitz DL, Friedman RA, Pyle AM. Catalytic role of 2′-hydroxyl groups within a group II intron active site. Science. 1996;271:1410–1413. - PubMed
-
- Adams PL, Stahley MR, Kosek AB, Wang J, Strobel SA. Crystal structure of a self-splicing group I intron with both exons. Nature. 2004;430:45–50. - PubMed
-
- Ban N, Nissen P, Hansen J, Moore PB, Steitz TA. The complete atomic structure of the large ribosomal subunit at 2.4 Å resolution. Science. 2000;289:905–920. - PubMed
-
- Batey RT, Gilbert SD, Montange RK. Structure of a natural guanine-responsive riboswitch complexecd with the metabolite hypoxanthine. Nature. 2004;432:411–15. - PubMed
-
- Beckers ML, Melssen WJ, Buydens LM. Predicting nucleic acid torsion angle values using artificial neural networks. Journal of Computer-Aided Molecular Design. 1998;12:53–61. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
