

Detection of inferred CCR5- and CXCR4-using HIV-1 variants and evolutionary intermediates using ultra-deep pyrosequencing
- PMID: 21731496
- PMCID: PMC3121885
- DOI: 10.1371/journal.ppat.1002106
Detection of inferred CCR5- and CXCR4-using HIV-1 variants and evolutionary intermediates using ultra-deep pyrosequencing
Abstract
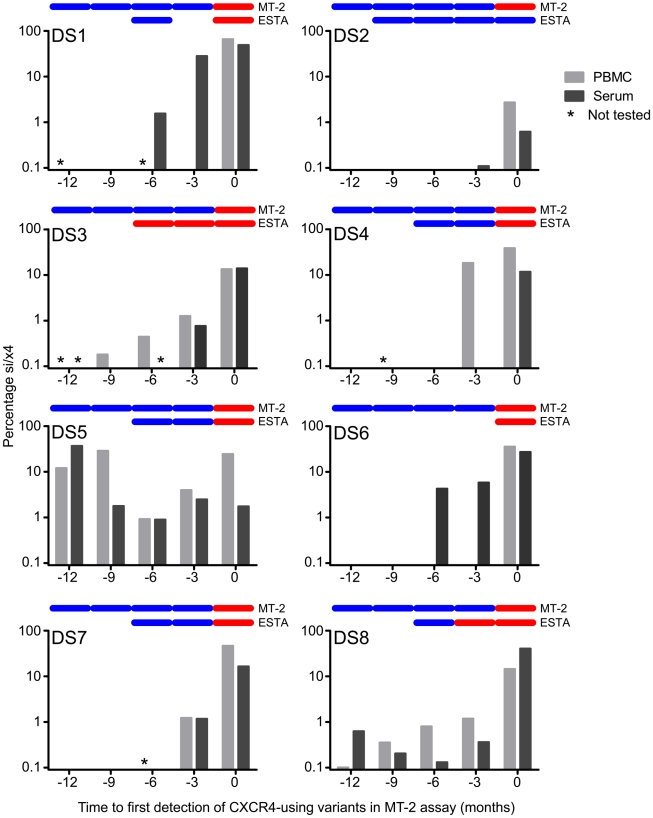
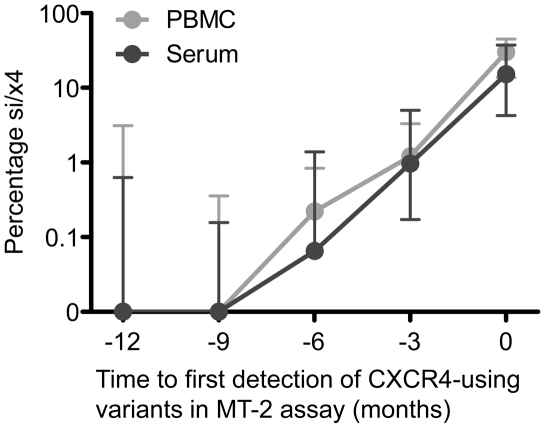
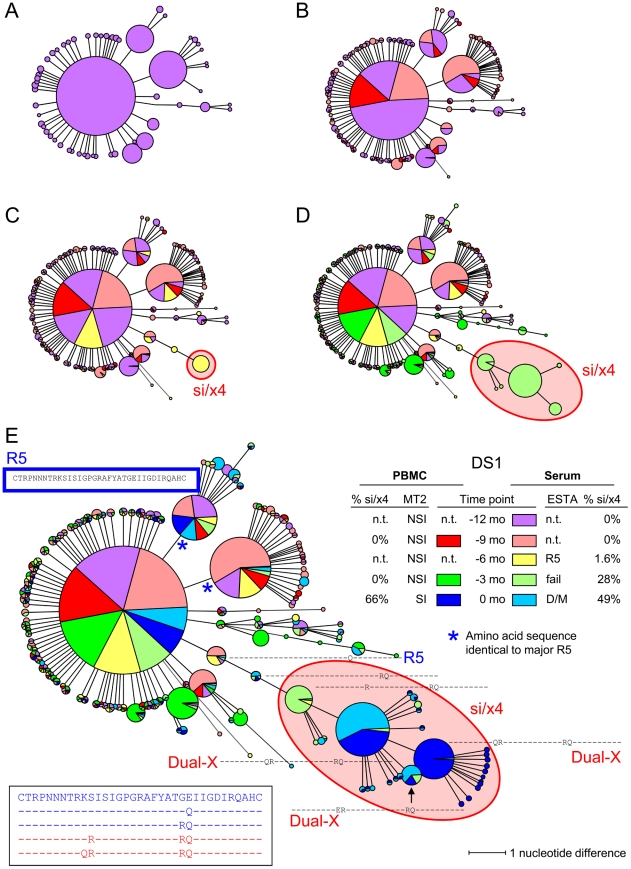
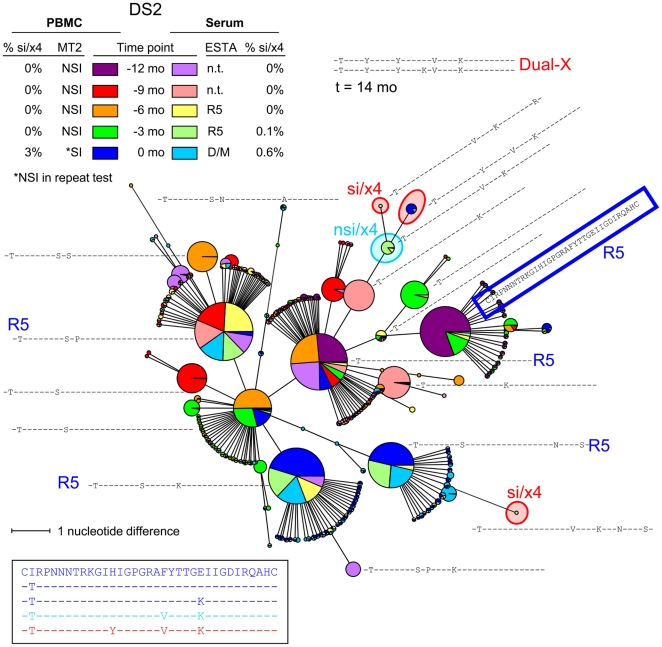
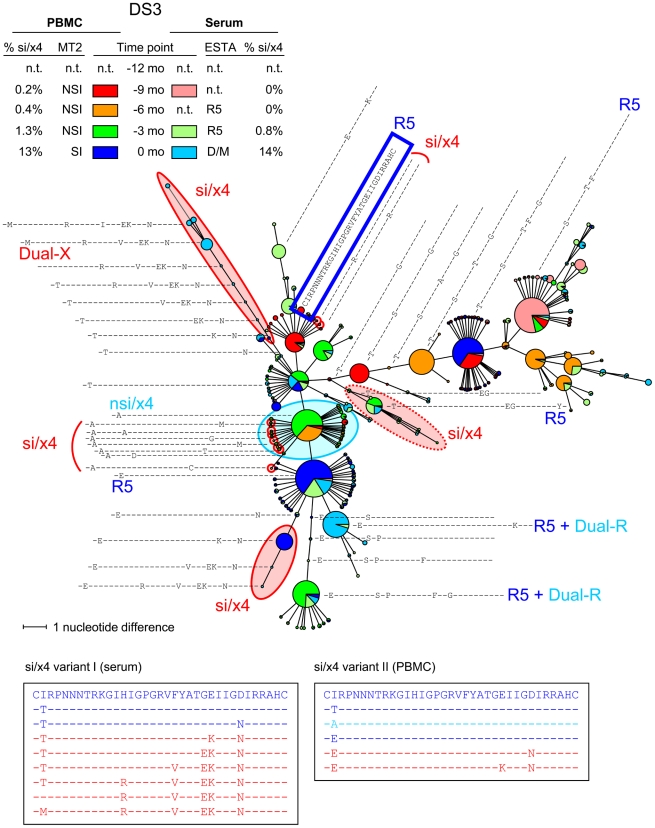
The emergence of CXCR4-using human immunodeficiency virus type 1 (HIV-1) variants is associated with accelerated disease progression. CXCR4-using variants are believed to evolve from CCR5-using variants, but due to the extremely low frequency at which transitional intermediate variants are often present, the kinetics and mutational pathways involved in this process have been difficult to study and are therefore poorly understood. Here, we used ultra-deep sequencing of the V3 loop of the viral envelope in combination with the V3-based coreceptor prediction tools PSSM(NSI/SI) and geno2pheno([coreceptor]) to detect HIV-1 variants during the transition from CCR5- to CXCR4-usage. We analyzed PBMC and serum samples obtained from eight HIV-1-infected individuals at three-month intervals up to one year prior to the first phenotypic detection of CXCR4-using variants in the MT-2 assay. Between 3,482 and 10,521 reads were generated from each sample. In all individuals, V3 sequences of predicted CXCR4-using HIV-1 were detected at least three months prior to phenotypic detection of CXCR4-using variants in the MT-2 assay. Subsequent analysis of the genetic relationships of these V3 sequences using minimum spanning trees revealed that the transition in coreceptor usage followed a stepwise mutational pathway involving sequential intermediate variants, which were generally present at relatively low frequencies compared to the major predicted CCR5- and CXCR4-using variants. In addition, we observed differences between individuals with respect to the number of predicted CXCR4-using variants, the diversity among major predicted CCR5-using variants, and the presence or absence of intermediate variants with discordant phenotype predictions. These results provide the first detailed description of the mutational pathways in V3 during the transition from CCR5- to CXCR4-usage in natural HIV-1 infection.
Conflict of interest statement
I have read the journal's policy and have the following conflicts. WH, AF, CJP and EC are employees of Monogram Biosciences. PRH is a consultant for and/or has received grant funding from Merck, ViiV Healthcare, Pfizer, Quest, Virco and Abbott.
Figures
References
-
- Zhu T, Mo H, Wang N, Nam DS, Cao Y, et al. Genotypic and phenotypic characterization of HIV-1 in patients with primary infection. Science. 1993;261:1179–1181. - PubMed
-
- Schuitemaker H, Koot M, Kootstra NA, Dercksen MW, De Goede REY, et al. Biological phenotype of human immunodeficiency virus type 1 clones at different stages of infection: progression of disease is associated with a shift from monocytotropic to T-cell-tropic virus populations. J Virol. 1992;66:1354–1360. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
