

An information theoretic, microfluidic-based single cell analysis permits identification of subpopulations among putatively homogeneous stem cells
- PMID: 21731674
- PMCID: PMC3120839
- DOI: 10.1371/journal.pone.0021211
An information theoretic, microfluidic-based single cell analysis permits identification of subpopulations among putatively homogeneous stem cells
Abstract
An incomplete understanding of the nature of heterogeneity within stem cell populations remains a major impediment to the development of clinically effective cell-based therapies. Transcriptional events within a single cell are inherently stochastic and can produce tremendous variability, even among genetically identical cells. It remains unclear how mammalian cellular systems overcome this intrinsic noisiness of gene expression to produce consequential variations in function, and what impact this has on the biologic and clinical relevance of highly 'purified' cell subgroups. To address these questions, we have developed a novel method combining microfluidic-based single cell analysis and information theory to characterize and predict transcriptional programs across hundreds of individual cells. Using this technique, we demonstrate that multiple subpopulations exist within a well-studied and putatively homogeneous stem cell population, murine long-term hematopoietic stem cells (LT-HSCs). These subgroups are defined by nonrandom patterns that are distinguishable from noise and are consistent with known functional properties of these cells. We anticipate that this analytic framework can also be applied to other cell types to elucidate the relationship between transcriptional and phenotypic variation.
Conflict of interest statement
Figures
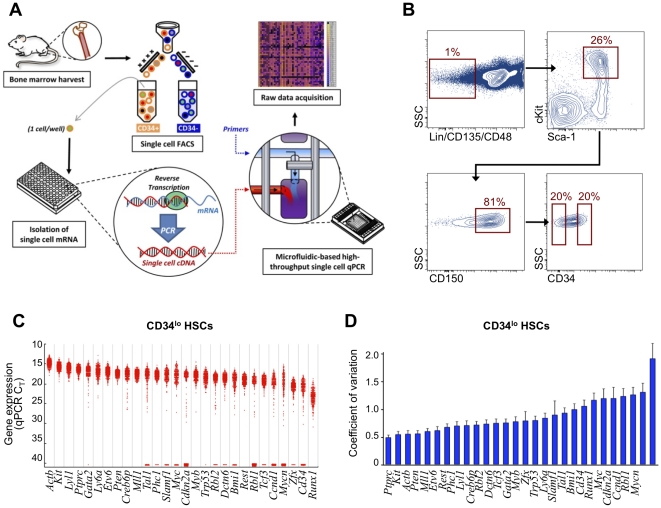
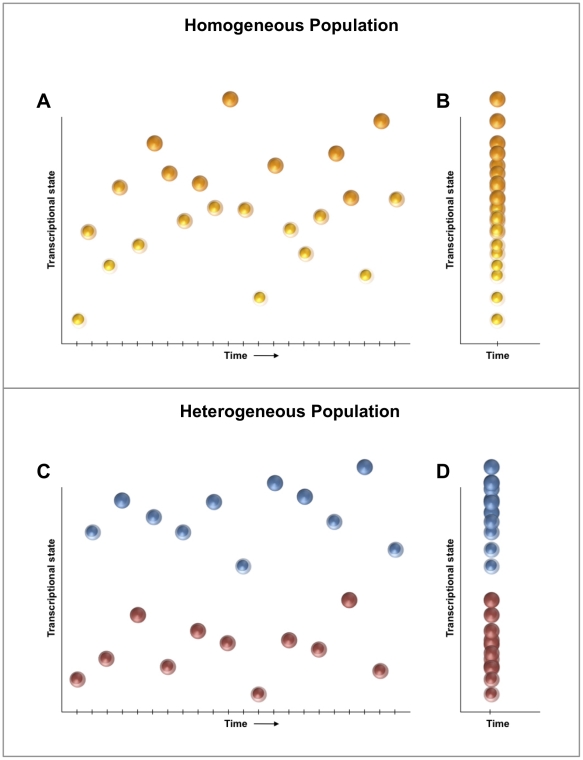
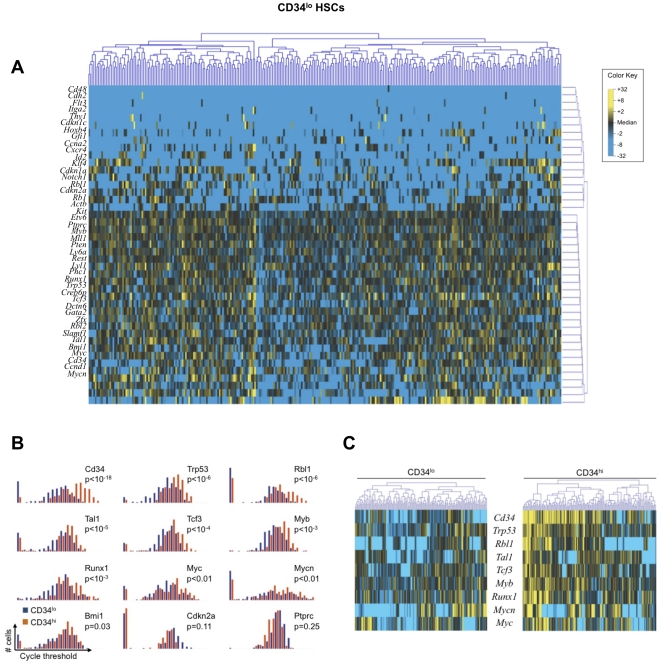
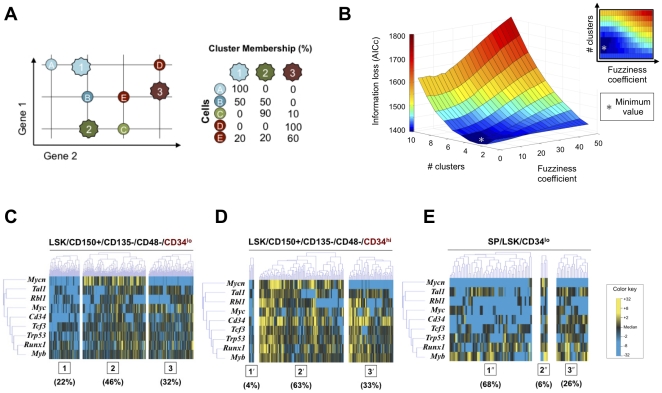
References
-
- Bar-Even A, Paulsson J, Maheshri N, Carmi M, O'Shea E, et al. Noise in protein expression scales with natural protein abundance. Nat Genet. 2006;38:636–643. - PubMed
-
- Elowitz MB, Levine AJ, Siggia ED, Swain PS. Stochastic gene expression in a single cell. SCIENCE. 2002;297:1183–1186. - PubMed
-
- Levsky JM, Shenoy SM, Pezo RC, Singer RH. Single-cell gene expression profiling. SCIENCE. 2002;297:836–840. - PubMed
-
- Ozbudak EM, Thattai M, Kurtser I, Grossman AD, van Oudenaarden A. Regulation of noise in the expression of a single gene. Nat Genet. 2002;31:69–73. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
