

A novel missense mutation in the connexin30 causes nonsyndromic hearing loss
- PMID: 21731760
- PMCID: PMC3123352
- DOI: 10.1371/journal.pone.0021473
A novel missense mutation in the connexin30 causes nonsyndromic hearing loss
Abstract
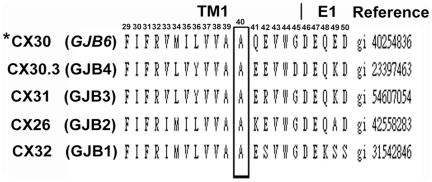
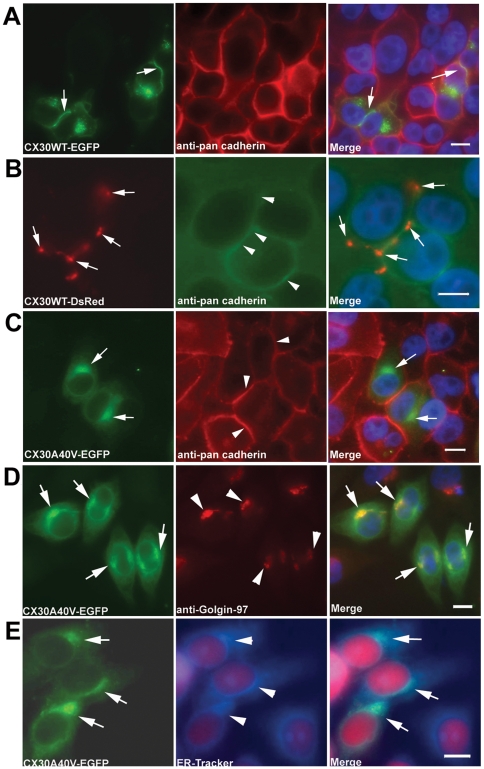
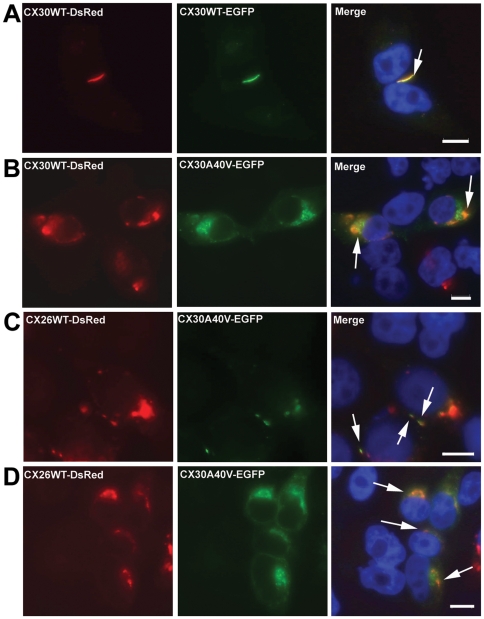
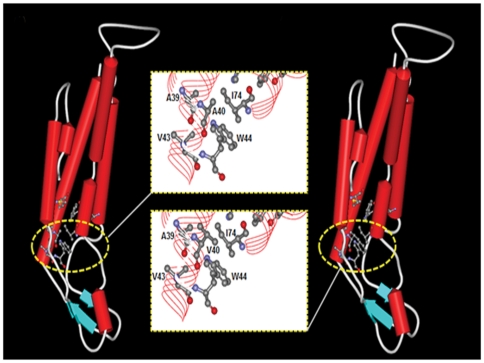
Dysfunctional gap junctions caused by GJB2 (CX26) and GJB6 (CX30) mutations are implicated in nearly half of nonsyndromic hearing loss cases. A recent study identified a heterozygous mutation, c.119C>T (p.A40V), in the GJB6 gene of patients with nonsyndromic hearing loss. However, the functional role of the mutation in hearing loss remains unclear. In this study, analyses of cell biology indicated that a p.A40V missense mutation of CX30 causes CX30 protein accumulation in the Golgi body rather than in the cytoplasmic membrane. The tet-on protein expression system was used for further study of mutant proteins in CX30 and CX30A40V co-expressions and in CX26 and CX30A40V co-expressions. The p.A40V missense mutation exerted a dominant negative effect on both normal CX30 and CX26, which impaired gap junction formation. Moreover, computer-assisted modeling suggested that this p.A40V mutation affects the intra molecular interaction in the hydrophobic core of Trp44, which significantly alters the efficiency of gap junction formation. These findings suggest that the p.A40V mutation in CX30 causes autosomal-dominant nonsyndromic hearing loss. These data provide a novel molecular explanation for the role of GJB6 in hearing loss.
Conflict of interest statement
Figures
References
-
- Petit C. Genes responsible for human hereditary deafness: symphony of a thousand. Nature Genet. 1996;14:385–391. - PubMed
-
- Apps SA, Rankin WA, Kurmis AP. Connexin26 mutations in autosomal recessive deafness disorders: A review. Int J Audiol. 2007;46:75–81. - PubMed
-
- Morton NE. Genetic epidemiogy of hearing impairment. Ann NY Acad Sci. 1991;630:16–31. - PubMed
-
- Pallares-Ruiz N, Blanchet P, Mondain M, Claustres M, Roux AF. A large deletion including most of GJB6 in recessive nonsyndromic deafness: a digenic effect? Eur J Hum Genet. 2002;10:72–76. - PubMed
-
- Beyer EC, Berthoud VM. Harris A, Locke D, editors. The family of connexin gene. Connexin: A Guide. 2009. pp. 3–26. Humana Press, Springer, USA.
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Miscellaneous
