

Pathogenic forms of tau inhibit kinesin-dependent axonal transport through a mechanism involving activation of axonal phosphotransferases
- PMID: 21734277
- PMCID: PMC3391724
- DOI: 10.1523/JNEUROSCI.0560-11.2011
Pathogenic forms of tau inhibit kinesin-dependent axonal transport through a mechanism involving activation of axonal phosphotransferases
Abstract
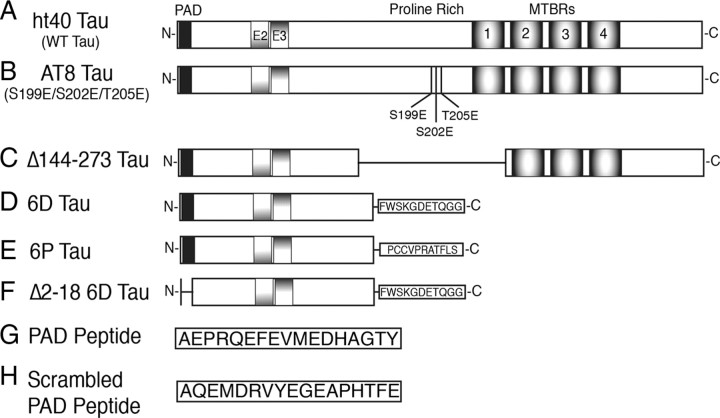
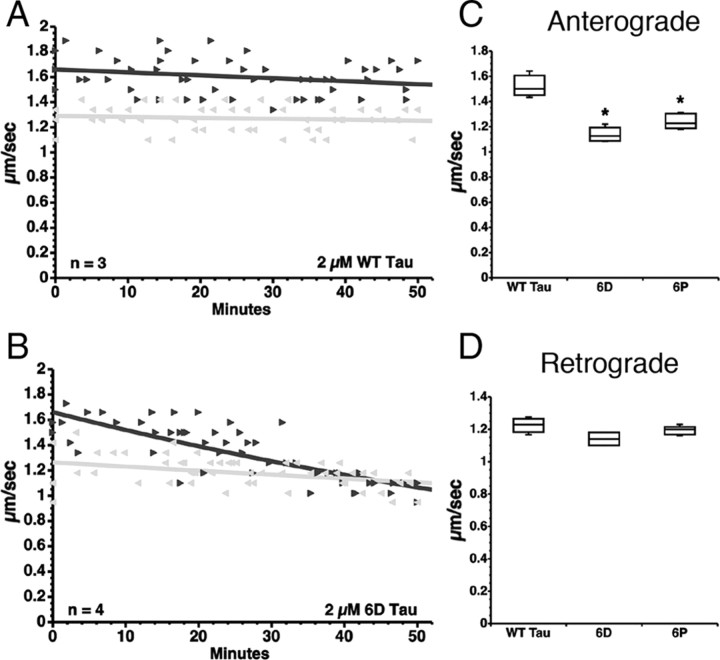
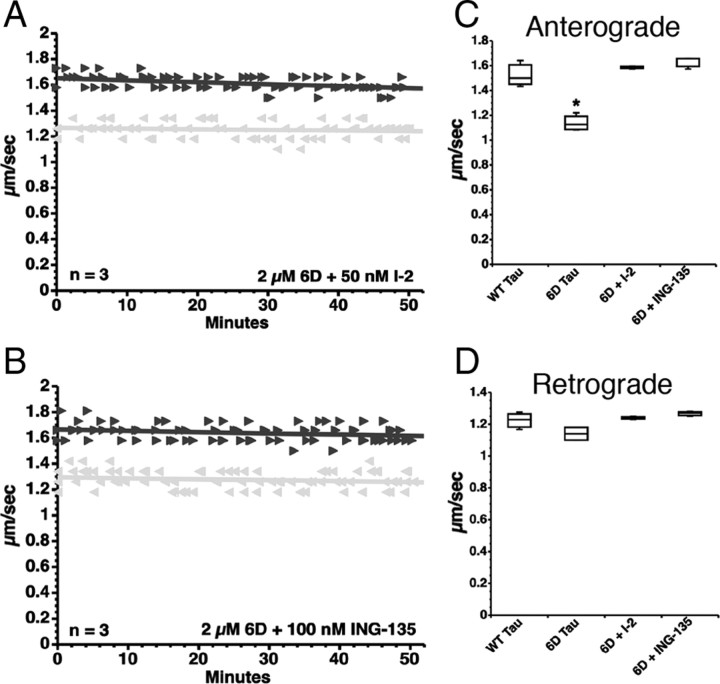
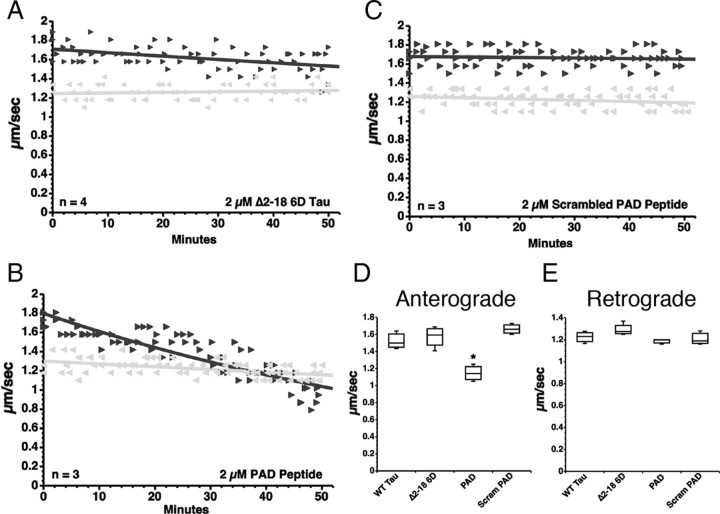
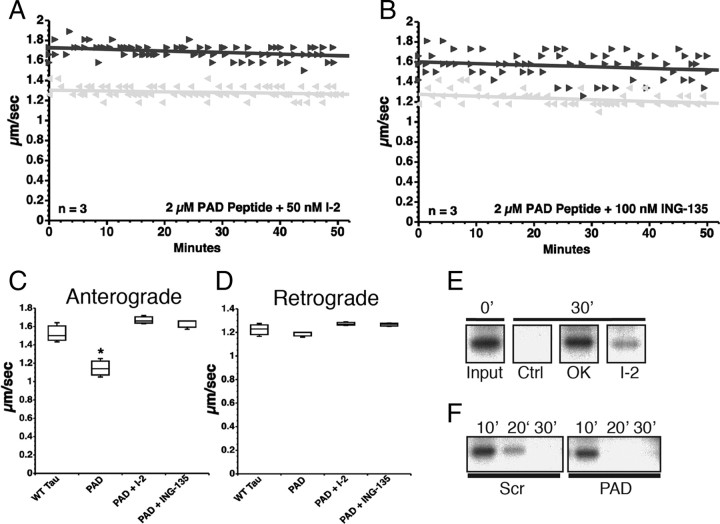
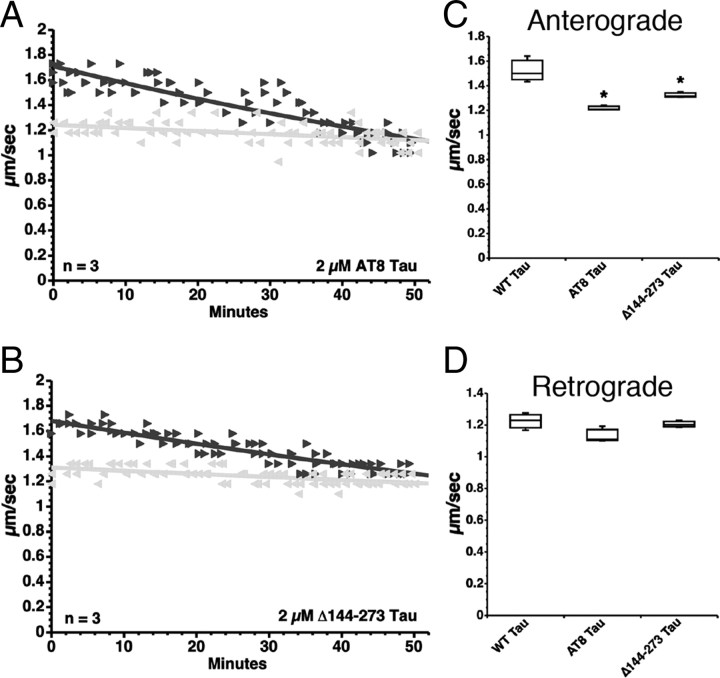
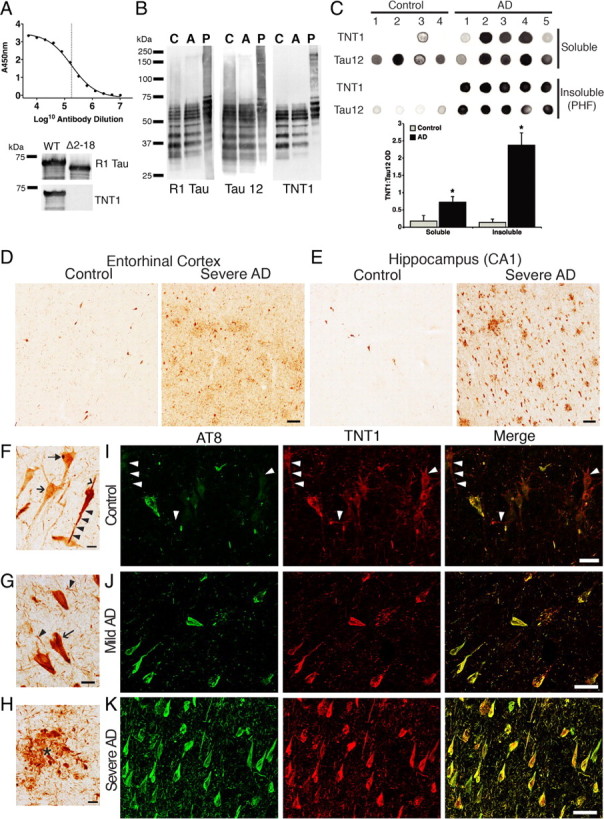
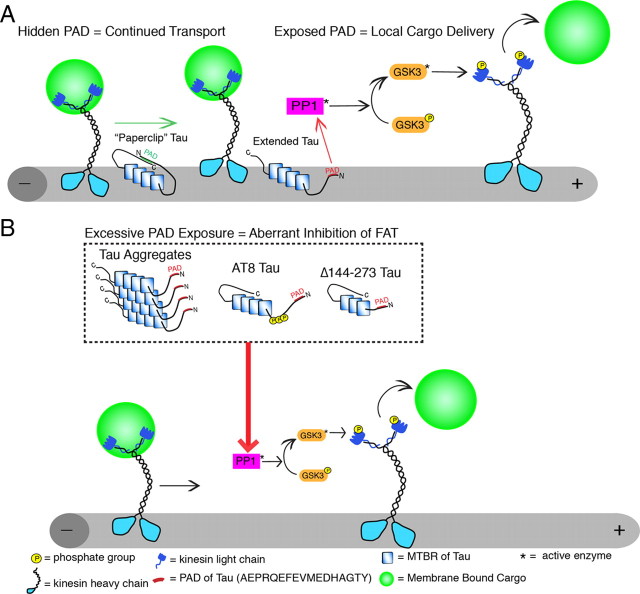
Aggregated filamentous forms of hyperphosphorylated tau (a microtubule-associated protein) represent pathological hallmarks of Alzheimer's disease (AD) and other tauopathies. While axonal transport dysfunction is thought to represent a primary pathogenic factor in AD and other neurodegenerative diseases, the direct molecular link between pathogenic forms of tau and deficits in axonal transport remain unclear. Recently, we demonstrated that filamentous, but not soluble, forms of wild-type tau inhibit anterograde, kinesin-based fast axonal transport (FAT) by activating axonal protein phosphatase 1 (PP1) and glycogen synthase kinase 3 (GSK3), independent of microtubule binding. Here, we demonstrate that amino acids 2-18 of tau, comprising a phosphatase-activating domain (PAD), are necessary and sufficient for activation of this pathway in axoplasms isolated from squid giant axons. Various pathogenic forms of tau displaying increased exposure of PAD inhibited anterograde FAT in squid axoplasm. Importantly, immunohistochemical studies using a novel PAD-specific monoclonal antibody in human postmortem tissue indicated that increased PAD exposure represents an early pathogenic event in AD that closely associates in time with AT8 immunoreactivity, an early marker of pathological tau. We propose a model of pathogenesis in which disease-associated changes in tau conformation lead to increased exposure of PAD, activation of PP1-GSK3, and inhibition of FAT. Results from these studies reveal a novel role for tau in modulating axonal phosphotransferases and provide a molecular basis for a toxic gain-of-function associated with pathogenic forms of tau.
Figures
References
-
- Alonso AC, Grundke-Iqbal I, Iqbal K. Alzheimer's disease hyperphosphorylated tau sequesters normal tau into tangles of filaments and disassembles microtubules. Nat Med. 1996;2:783–787. - PubMed
-
- Andreadis A. Tau gene alternative splicing: expression patterns, regulation and modulation of function in normal brain and neurodegenerative diseases. Biochim Biophys Acta. 2005;1739:91–103. - PubMed
-
- Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer's disease. Neurology. 1992;42:631–639. - PubMed
-
- Barghorn S, Davies P, Mandelkow E. Tau paired helical filaments from Alzheimer's disease brain and assembled in vitro are based on beta-structure in the core domain. Biochemistry. 2004;43:1694–1703. - PubMed
-
- Berry RW, Sweet AP, Clark FA, Lagalwar S, Lapin BR, Wang T, Topgi S, Guillozet-Bongaarts AL, Cochran EJ, Bigio EH, Binder LI. Tau epitope display in progressive supranuclear palsy and corticobasal degeneration. J Neurocytol. 2004;33:287–295. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources