

Phylogenetic analysis consistent with a clinical history of sexual transmission of HIV-1 from a single donor reveals transmission of highly distinct variants
- PMID: 21736738
- PMCID: PMC3161944
- DOI: 10.1186/1742-4690-8-54
Phylogenetic analysis consistent with a clinical history of sexual transmission of HIV-1 from a single donor reveals transmission of highly distinct variants
Abstract
Background: To combat the pandemic of human immunodeficiency virus 1 (HIV-1), a successful vaccine will need to cope with the variability of transmissible viruses. Human hosts infected with HIV-1 potentially harbour many viral variants but very little is known about viruses that are likely to be transmitted, or even if there are viral characteristics that predict enhanced transmission in vivo. We show for the first time that genetic divergence consistent with a single transmission event in vivo can represent several years of pre-transmission evolution.
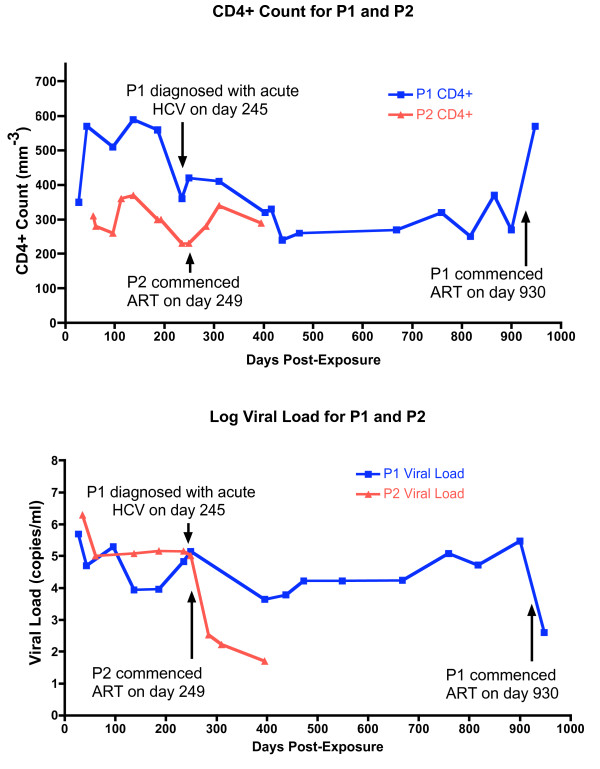
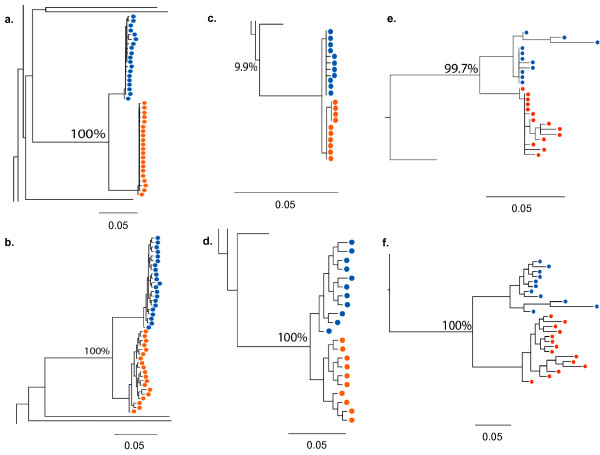
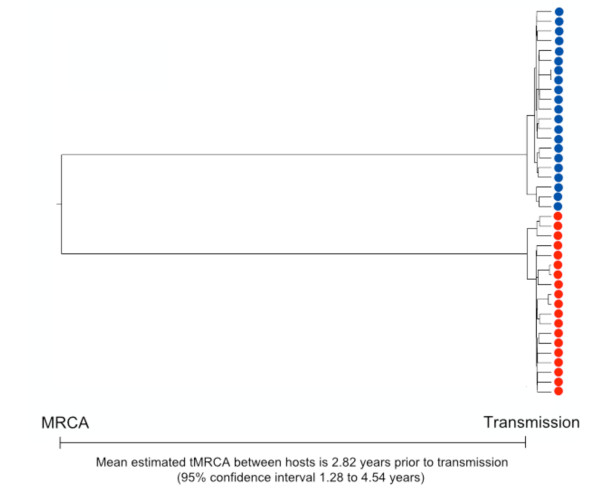
Results: We describe a highly unusual case consistent with a single donor transmitting highly related but distinct HIV-1 variants to two individuals on the same evening. We confirm that the clustering of viral genetic sequences, present within each recipient, is consistent with the history of a single donor across the viral env, gag and pol genes by maximum likelihood and bayesian Markov Chain Monte Carlo based phylogenetic analyses. Based on an uncorrelated, lognormal relaxed clock of env gene evolution calibrated with other datasets, the time since the most recent common ancestor is estimated as 2.86 years prior to transmission (95% confidence interval 1.28 to 4.54 years).
Conclusion: Our results show that an effective design for a preventative vaccine will need to anticipate extensive HIV-1 diversity within an individual donor as well as diversity at the population level.
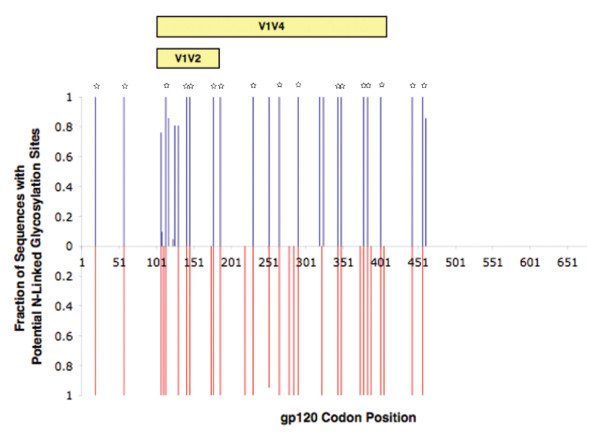
Figures
References
-
- Salazar-Gonzalez JF, Salazar MG, Keele BF, Learn GH, Giorgi EE, Li H, Decker JM, Wang S, Baalwa J, Kraus MH. et al. Genetic identity, biological phenotype, and evolutionary pathways of transmitted/founder viruses in acute and early HIV-1 infection. J Exp Med. 2009;206:1273–1289. doi: 10.1084/jem.20090378. - DOI - PMC - PubMed
-
- Salazar-Gonzalez JF, Bailes E, Pham KT, Salazar MG, Guffey MB, Keele BF, Derdeyn CA, Farmer P, Hunter E, Allen S. et al. Deciphering human immunodeficiency virus type 1 transmission and early envelope diversification by single-genome amplification and sequencing. J Virol. 2008;82:3952–3970. doi: 10.1128/JVI.02660-07. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
