

G protein-dependent and G protein-independent signaling pathways and their impact on cardiac function
- PMID: 21737817
- PMCID: PMC3138127
- DOI: 10.1161/CIRCRESAHA.110.231225
G protein-dependent and G protein-independent signaling pathways and their impact on cardiac function
Abstract
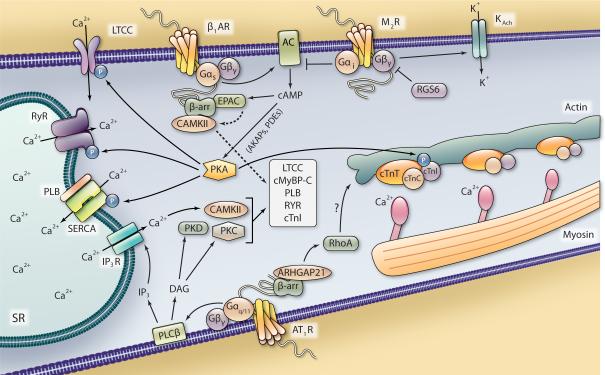
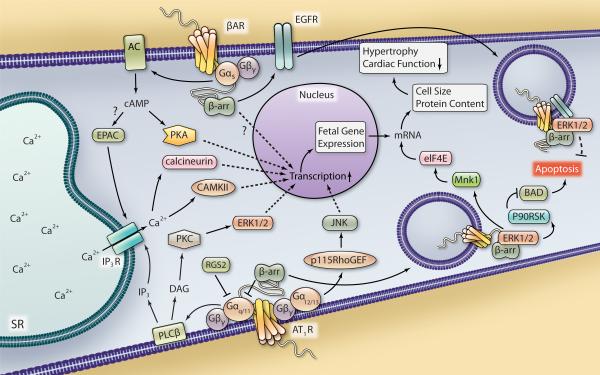
G protein-coupled receptors signal through a variety of mechanisms that impact cardiac function, including contractility and hypertrophy. G protein-dependent and G protein-independent pathways each have the capacity to initiate numerous intracellular signaling cascades to mediate these effects. G protein-dependent signaling has been studied for decades and great strides continue to be made in defining the intricate pathways and effectors regulated by G proteins and their impact on cardiac function. G protein-independent signaling is a relatively newer concept that is being explored more frequently in the cardiovascular system. Recent studies have begun to reveal how cardiac function may be regulated via G protein-independent signaling, especially with respect to the ever-expanding cohort of β-arrestin-mediated processes. This review primarily focuses on the impact of both G protein-dependent and β-arrestin-dependent signaling pathways on cardiac function, highlighting the most recent data that illustrate the comprehensive nature of these mechanisms of G protein-coupled receptor signaling.
Figures
References
-
- Penela P, Murga C, Ribas C, Tutor AS, Peregrin S, Mayor F., Jr. Mechanisms of regulation of G protein-coupled receptor kinases (GRKs) and cardiovascular disease. Cardiovasc Res. 2006;69:46–56. - PubMed
-
- Tilley DG, Rockman HA. Role of beta-adrenergic receptor signaling and desensitization in heart failure: new concepts and prospects for treatment. Expert Rev Cardiovasc Ther. 2006;4:417–432. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
