

The immunology of stroke: from mechanisms to translation
- PMID: 21738161
- PMCID: PMC3137275
- DOI: 10.1038/nm.2399
The immunology of stroke: from mechanisms to translation
Abstract
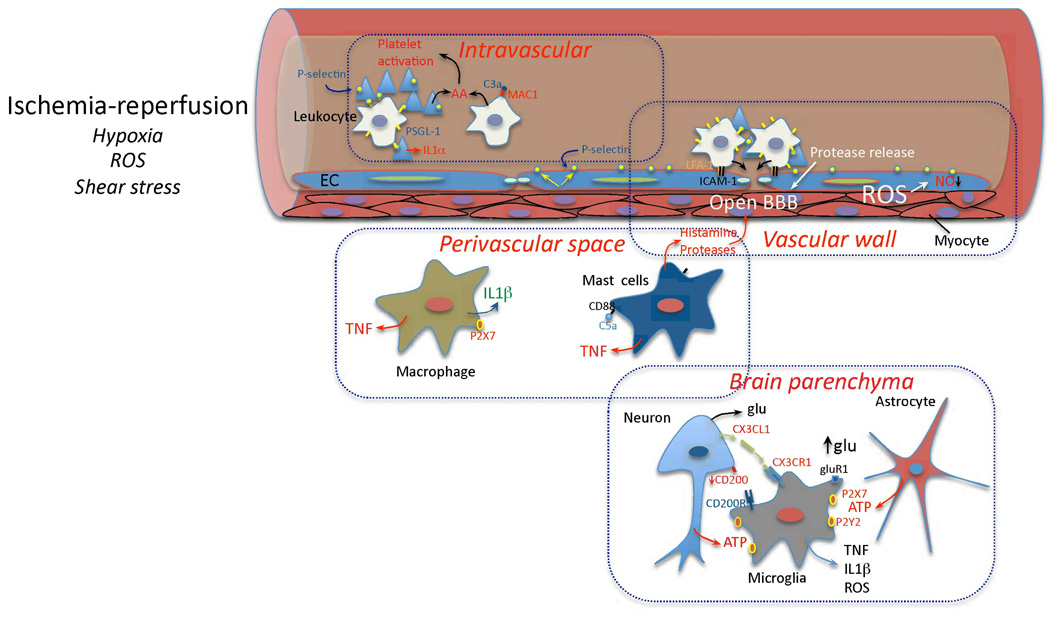
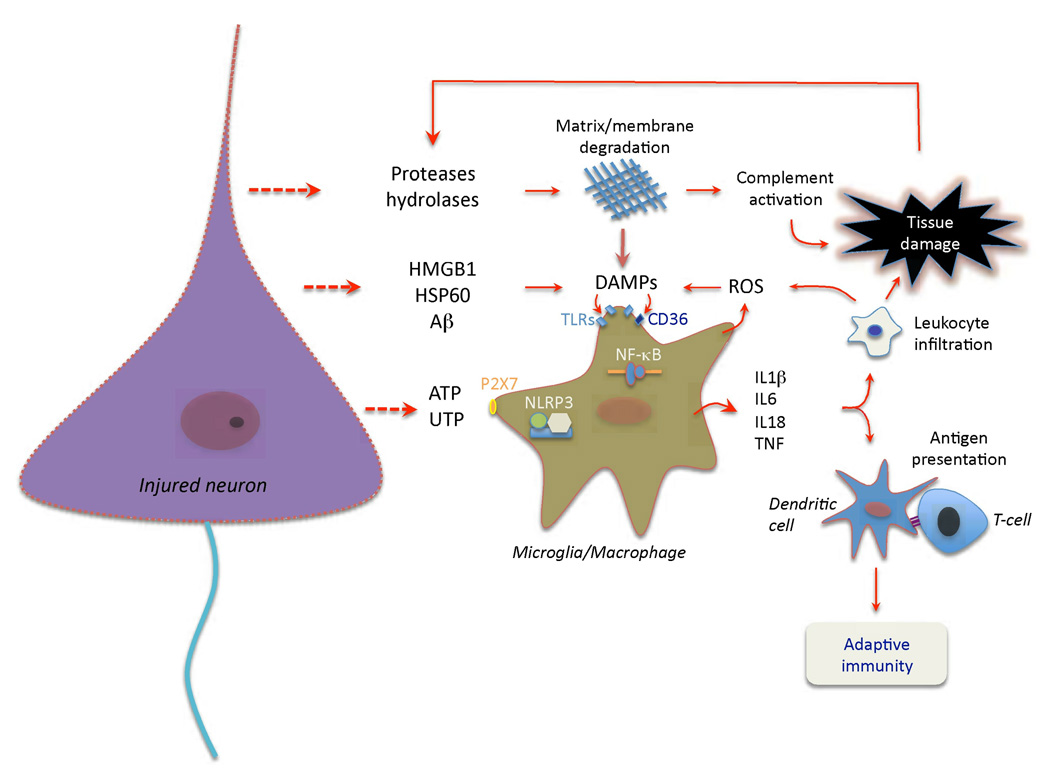
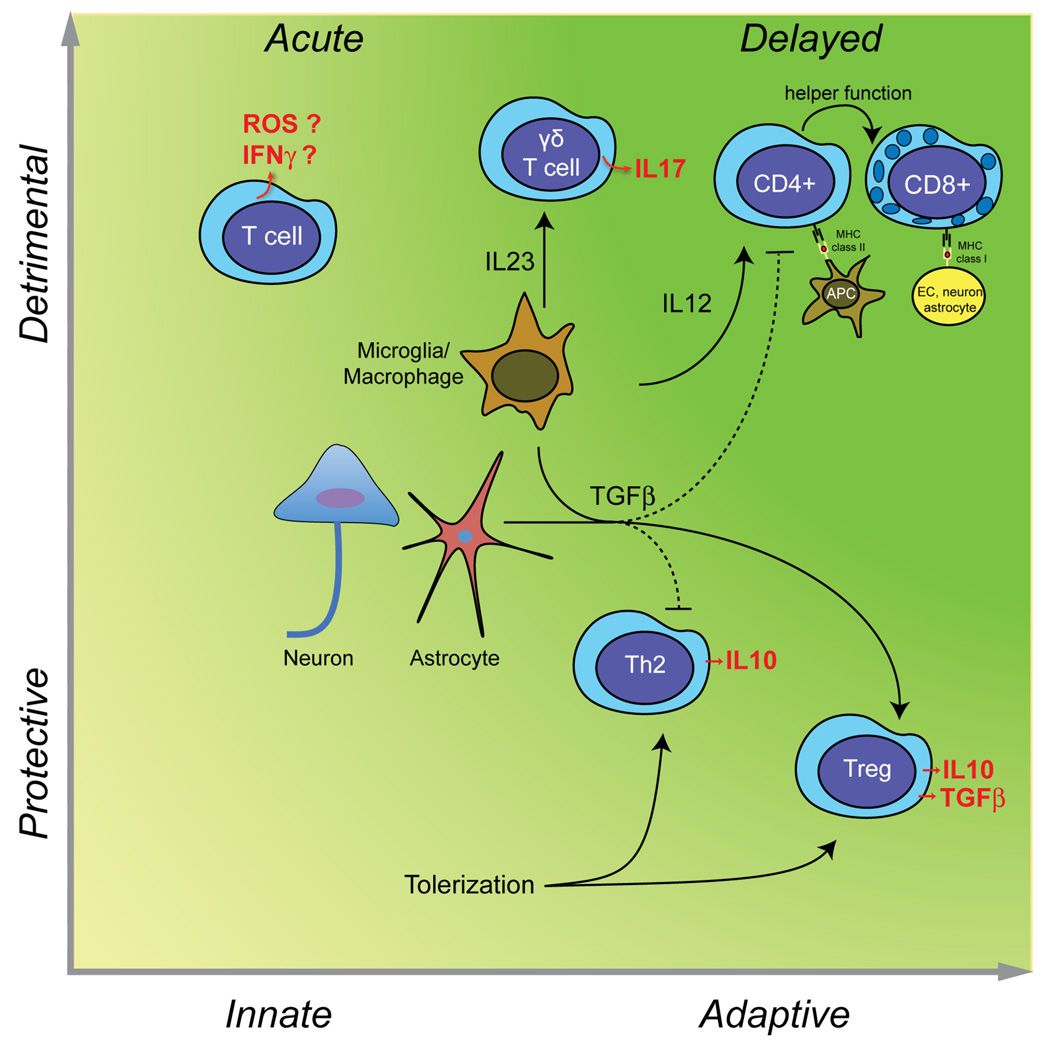
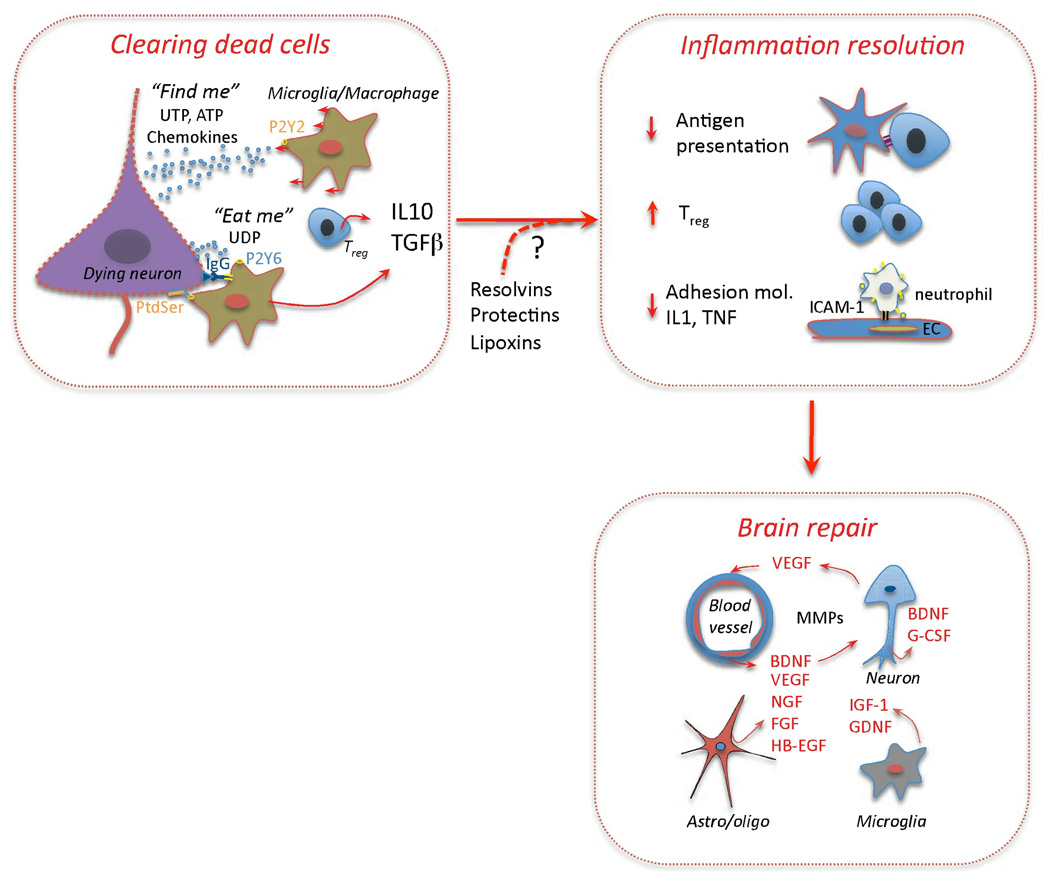
Immunity and inflammation are key elements of the pathobiology of stroke, a devastating illness second only to cardiac ischemia as a cause of death worldwide. The immune system participates in the brain damage produced by ischemia, and the damaged brain, in turn, exerts an immunosuppressive effect that promotes fatal infections that threaten the survival of people after stroke. Inflammatory signaling is involved in all stages of the ischemic cascade, from the early damaging events triggered by arterial occlusion to the late regenerative processes underlying post-ischemic tissue repair. Recent developments have revealed that stroke engages both innate and adaptive immunity. But adaptive immunity triggered by newly exposed brain antigens does not have an impact on the acute phase of the damage. Nevertheless, modulation of adaptive immunity exerts a remarkable protective effect on the ischemic brain and offers the prospect of new stroke therapies. As immunomodulation is not devoid of deleterious side effects, a better understanding of the reciprocal interaction between the immune system and the ischemic brain is essential to harness the full therapeutic potential of the immunology of stroke.
Figures
Comment in
-
Mannose-binding lectin-the forgotten molecule?Nat Med. 2011 Dec 6;17(12):1547-8; author reply 1548. doi: 10.1038/nm.2588. Nat Med. 2011. PMID: 22146452 No abstract available.
References
-
- Mena H, Cadavid D, Rushing EJ. Human cerebral infarct: a proposed histopathologic classification based on 137 cases. Acta Neuropathol. 2004;108:524–530. - PubMed
-
- Meisel C, Schwab J, Prass K, Meisel A, Dirnagl U. Central nervous system injury-induced immune deficiency syndrome. Nat Rev Neurosci. 2005;6:775–786. - PubMed
-
- Urra X, Cervera A, Villamor N, Planas AM, Chamorro A. Harms and benefits of lymphocyte subpopulations in patients with acute stroke. Neuroscience. 2009;158:1174–1183. - PubMed
-
- Weiner HL. The challenge of multiple sclerosis: how do we cure a chronic heterogeneous disease? Ann Neurol. 2009;65:239–248. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
