

A frameshift mutation in golden retriever dogs with progressive retinal atrophy endorses SLC4A3 as a candidate gene for human retinal degenerations
- PMID: 21738669
- PMCID: PMC3124514
- DOI: 10.1371/journal.pone.0021452
A frameshift mutation in golden retriever dogs with progressive retinal atrophy endorses SLC4A3 as a candidate gene for human retinal degenerations
Abstract
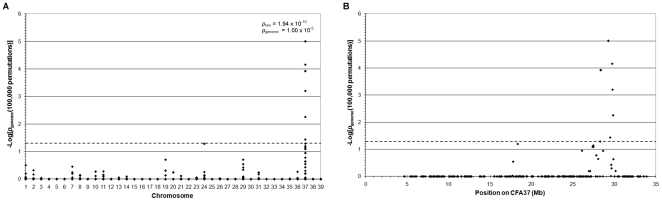
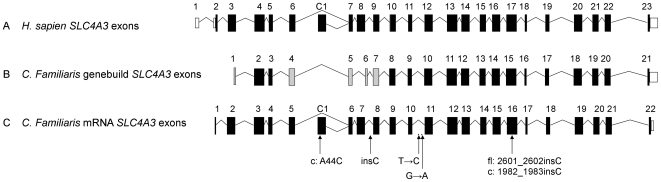
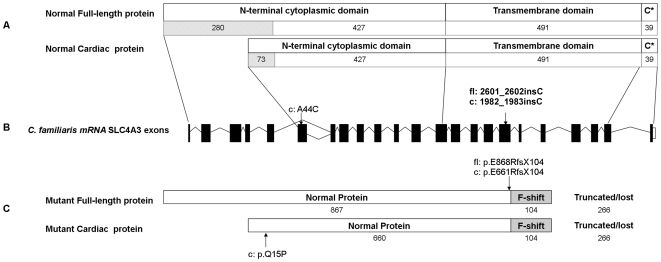
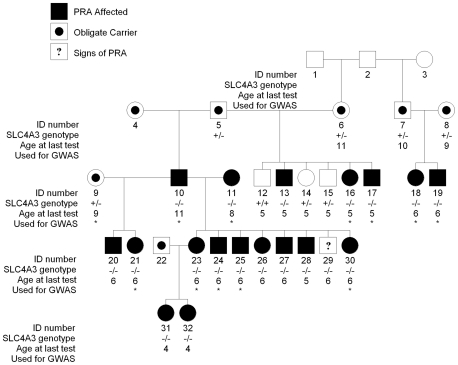
Progressive retinal atrophy (PRA) in dogs, the canine equivalent of retinitis pigmentosa (RP) in humans, is characterised by vision loss due to degeneration of the photoreceptor cells in the retina, eventually leading to complete blindness. It affects more than 100 dog breeds, and is caused by numerous mutations. RP affects 1 in 4000 people in the Western world and 70% of causal mutations remain unknown. Canine diseases are natural models for the study of human diseases and are becoming increasingly useful for the development of therapies in humans. One variant, prcd-PRA, only accounts for a small proportion of PRA cases in the Golden Retriever (GR) breed. Using genome-wide association with 27 cases and 19 controls we identified a novel PRA locus on CFA37 (p(raw) = 1.94×10(-10), p(genome) = 1.0×10(-5)), where a 644 kb region was homozygous within cases. A frameshift mutation was identified in a solute carrier anion exchanger gene (SLC4A3) located within this region. This variant was present in 56% of PRA cases and 87% of obligate carriers, and displayed a recessive mode of inheritance with full penetrance within those lineages in which it segregated. Allele frequencies are approximately 4% in the UK, 6% in Sweden and 2% in France, but the variant has not been found in GRs from the US. A large proportion of cases (approximately 44%) remain unexplained, indicating that PRA in this breed is genetically heterogeneous and caused by at least three mutations. SLC4A3 is important for retinal function and has not previously been associated with spontaneously occurring retinal degenerations in any other species, including humans.
Conflict of interest statement
Figures

Similar articles
-
Late-onset progressive retinal atrophy in the Gordon and Irish Setter breeds is associated with a frameshift mutation in C2orf71.Anim Genet. 2013 Apr;44(2):169-77. doi: 10.1111/j.1365-2052.2012.02379.x. Epub 2012 Jun 12. Anim Genet. 2013. PMID: 22686255
-
A putative silencer variant in a spontaneous canine model of retinitis pigmentosa.PLoS Genet. 2020 Mar 9;16(3):e1008659. doi: 10.1371/journal.pgen.1008659. eCollection 2020 Mar. PLoS Genet. 2020. PMID: 32150541 Free PMC article.
-
A novel mutation in TTC8 is associated with progressive retinal atrophy in the golden retriever.Canine Genet Epidemiol. 2014 Apr 16;1:4. doi: 10.1186/2052-6687-1-4. eCollection 2014. Canine Genet Epidemiol. 2014. PMID: 26401321 Free PMC article.
-
Natural models for retinitis pigmentosa: progressive retinal atrophy in dog breeds.Hum Genet. 2019 May;138(5):441-453. doi: 10.1007/s00439-019-01999-6. Epub 2019 Mar 23. Hum Genet. 2019. PMID: 30904946 Review.
-
A review of research to elucidate the causes of the generalized progressive retinal atrophies.Vet J. 1998 Jan;155(1):5-18. doi: 10.1016/s1090-0233(98)80028-2. Vet J. 1998. PMID: 9455155 Review.
Cited by
-
Hemangiosarcoma in dogs as a potential non-rodent animal model for drug discovery research of angiosarcoma in humans.Front Oncol. 2023 Dec 7;13:1250766. doi: 10.3389/fonc.2023.1250766. eCollection 2023. Front Oncol. 2023. PMID: 38130992 Free PMC article. Review.
-
Genetic and phenotypic variations of inherited retinal diseases in dogs: the power of within- and across-breed studies.Mamm Genome. 2012 Feb;23(1-2):40-61. doi: 10.1007/s00335-011-9361-3. Epub 2011 Nov 8. Mamm Genome. 2012. PMID: 22065099 Free PMC article. Review.
-
A Coding Variant in the Gene Bardet-Biedl Syndrome 4 (BBS4) Is Associated with a Novel Form of Canine Progressive Retinal Atrophy.G3 (Bethesda). 2017 Jul 5;7(7):2327-2335. doi: 10.1534/g3.117.043109. G3 (Bethesda). 2017. PMID: 28533336 Free PMC article.
-
Genome-wide analysis of parent-of-origin effects in non-syndromic orofacial clefts.Eur J Hum Genet. 2014 Jun;22(6):822-30. doi: 10.1038/ejhg.2013.235. Epub 2013 Oct 30. Eur J Hum Genet. 2014. PMID: 24169523 Free PMC article.
-
Whole Genome Sequencing of Giant Schnauzer Dogs with Progressive Retinal Atrophy Establishes NECAP1 as a Novel Candidate Gene for Retinal Degeneration.Genes (Basel). 2019 May 21;10(5):385. doi: 10.3390/genes10050385. Genes (Basel). 2019. PMID: 31117272 Free PMC article.
References
-
- Grondahl J. Estimation of prognosis and prevalence of retinitis pigmentosa and Usher syndrome in Norway. Clin Genet. 1987;31:255–264. - PubMed
-
- Haim M, Holm NV, Rosenberg T. Prevalence of retinitis pigmentosa and allied disorders in Denmark. I Main results. Acta Ophthalmol (Copenh) 1992;70:178–186. - PubMed
-
- Pagon RA. Retinitis pigmentosa. Surv Ophthalmol. 1988;33:137–177. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Research Materials
