

On the representability of complete genomes by multiple competing finite-context (Markov) models
- PMID: 21738720
- PMCID: PMC3128062
- DOI: 10.1371/journal.pone.0021588
On the representability of complete genomes by multiple competing finite-context (Markov) models
Abstract
A finite-context (Markov) model of order k yields the probability distribution of the next symbol in a sequence of symbols, given the recent past up to depth k. Markov modeling has long been applied to DNA sequences, for example to find gene-coding regions. With the first studies came the discovery that DNA sequences are non-stationary: distinct regions require distinct model orders. Since then, Markov and hidden Markov models have been extensively used to describe the gene structure of prokaryotes and eukaryotes. However, to our knowledge, a comprehensive study about the potential of Markov models to describe complete genomes is still lacking. We address this gap in this paper. Our approach relies on (i) multiple competing Markov models of different orders (ii) careful programming techniques that allow orders as large as sixteen (iii) adequate inverted repeat handling (iv) probability estimates suited to the wide range of context depths used. To measure how well a model fits the data at a particular position in the sequence we use the negative logarithm of the probability estimate at that position. The measure yields information profiles of the sequence, which are of independent interest. The average over the entire sequence, which amounts to the average number of bits per base needed to describe the sequence, is used as a global performance measure. Our main conclusion is that, from the probabilistic or information theoretic point of view and according to this performance measure, multiple competing Markov models explain entire genomes almost as well or even better than state-of-the-art DNA compression methods, such as XM, which rely on very different statistical models. This is surprising, because Markov models are local (short-range), contrasting with the statistical models underlying other methods, where the extensive data repetitions in DNA sequences is explored, and therefore have a non-local character.
Conflict of interest statement
Figures
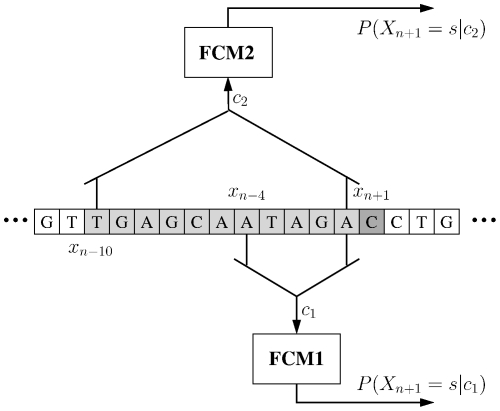
 and the context depths,
and the context depths,  , are
, are  and
and  . The probability of the next outcome,
. The probability of the next outcome,  , is conditioned by the last
, is conditioned by the last  outcomes. When more than one model is running competitively, the particular context depth used is chosen on a block basis.
outcomes. When more than one model is running competitively, the particular context depth used is chosen on a block basis.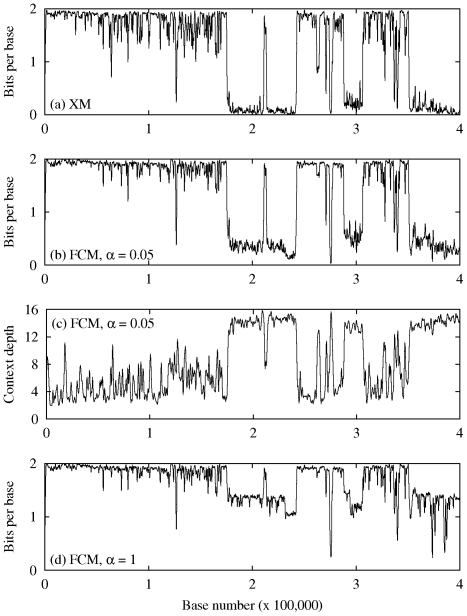
 for the high-order models (
for the high-order models ( ) and
) and  for the remainder models; (c) Variation of the depth of the context-model along the sequence, for the same setup as in (b); (d) The effect of parameter
for the remainder models; (c) Variation of the depth of the context-model along the sequence, for the same setup as in (b); (d) The effect of parameter  . In this case, we show the information sequence generated by the multiple competing finite-context models with
. In this case, we show the information sequence generated by the multiple competing finite-context models with  for all the models.
for all the models.References
-
- Grumbach S, Tahi F. Compression of DNA sequences. 1993. pp. 340–350. In: Proc. of the Data Compression Conf., DCC-93. Snowbird, Utah.
-
- Rivals E, Delahaye JP, Dauchet M, Delgrange O. A guaranteed compression scheme for repetitive DNA sequences. 1996. 453 In: Proc. of the Data Compression Conf., DCC-96. Snowbird, Utah.
-
- Loewenstern D, Yianilos PN. Significantly lower entropy estimates for natural DNA sequences. 1997. pp. 151–160. In: Proc. of the Data Compression Conf., DCC-97. Snowbird, Utah. - PubMed
-
- Chen X, Kwong S, Li M. A compression algorithm for DNA sequences. IEEE Engineering in Medicine and Biology Magazine. 2001;20:61–66. - PubMed
-
- Tabus I, Korodi G, Rissanen J. DNA sequence compression using the normalized maximum likelihood model for discrete regression. 2003. pp. 253–262. In: Proc. of the Data Compression Conf., DCC-2003. Snowbird, Utah.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
