

Melanoma-induced suppression of tumor antigen-specific T cell expansion is comparable to suppression of global T cell expansion
- PMID: 21741629
- PMCID: PMC4852956
- DOI: 10.1016/j.cellimm.2011.06.011
Melanoma-induced suppression of tumor antigen-specific T cell expansion is comparable to suppression of global T cell expansion
Abstract
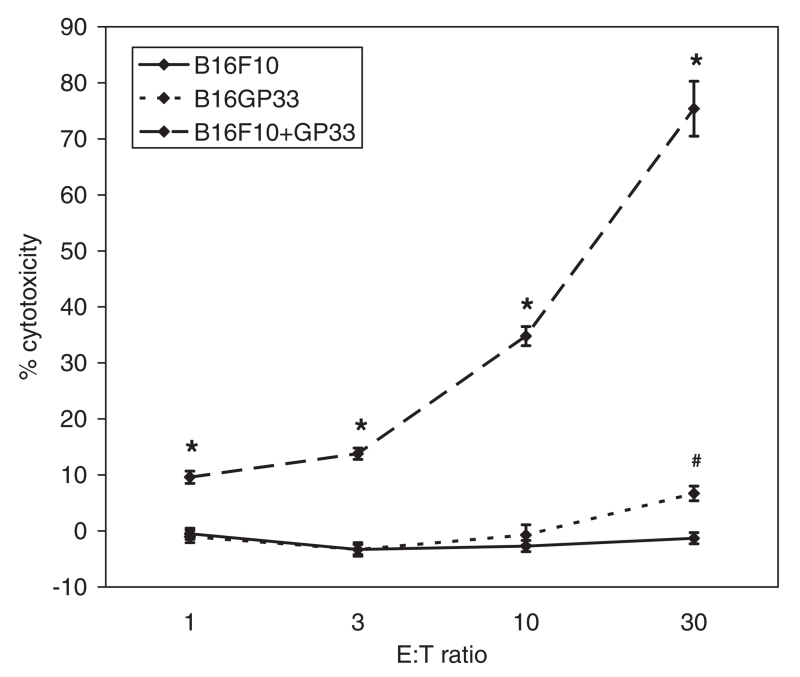
We have observed that in vivo interaction between melanoma and resting T cells promotes suppression of antigen-driven proliferative T cell expansion. We hypothesized that this suppression would affect tumor antigen-specific T cell populations more potently than tumor-unrelated T cell populations. A B16F10 cell line was stably transfected to express low levels of the lymphocytic choriomeningitis virus (LCMV) glycoprotein GP33 (B16GP33). Mice bearing B16F10 or B16GP33 tumors were infected with LCMV, and proliferative expansion of LCMV epitope-specific T cell populations was quantified. In vitro and in vivo assays confirmed low levels of antigenic GP33 expression by B16GP33 tumors. Suppressed expansion of GP33-specific T cells was equivalent between mice bearing B16F10 and B16GP33 tumors. These observations suggest that the ability of growing melanoma tumors to impair antigen-driven proliferative expansion of activated T cells is global and not antigen-specific, and provide further insight into the influence of cancer on activated T cell homeostasis.
Published by Elsevier Inc.
Figures





References
-
- Dunn GP, Old LJ, Schreiber RD. The three E’s of cancer immunoediting. Ann. Rev. Immunol. 2004;22:329–360. - PubMed
-
- Dunn GP, Old LJ, Schreiber RD. The immunobiology of cancer immunosurveillance and immunoediting. Immunity. 2004;21:137–148. - PubMed
-
- Shankaran V, Ikeda H, Bruce AT, et al. IFNγ and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature. 2001;410:1107–1111. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
