

Polarizable Simulations with Second order Interaction Model (POSSIM) force field: Developing parameters for alanine peptides and protein backbone
- PMID: 21743799
- PMCID: PMC3129858
- DOI: 10.1021/ct1007197
Polarizable Simulations with Second order Interaction Model (POSSIM) force field: Developing parameters for alanine peptides and protein backbone
Abstract
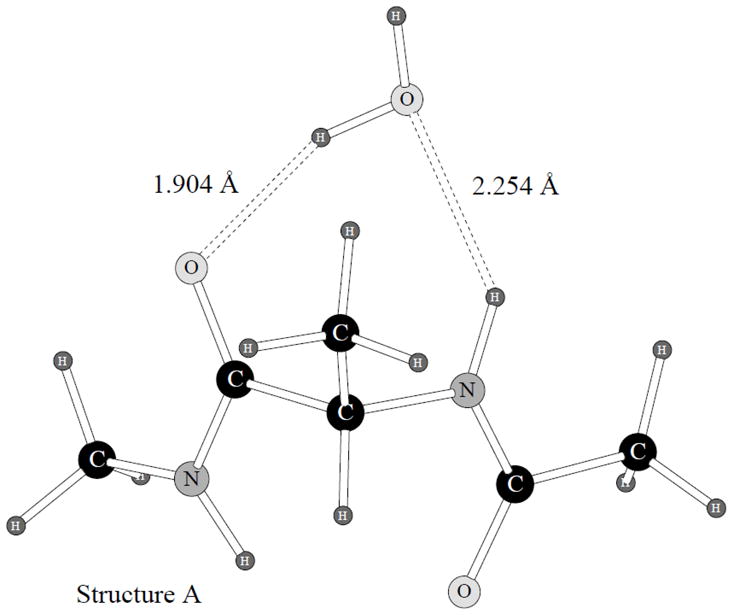
A previously introduced POSSIM (POlarizable Simulations with Second order Interaction Model) force field has been extended to include parameters for alanine peptides and protein backbones. New features were introduced into the fitting protocol, as compared to the previous generation of the polarizable force field for proteins. A reduced amount of quantum mechanical data was employed in fitting the electrostatic parameters. Transferability of the electrostatics between our recently developed NMA model and the protein backbone was confirmed. Binding energy and geometry for complexes of alanine dipeptide with a water molecule were estimated and found in a good agreement with high-level quantum mechanical results (for example, the intermolecular distances agreeing within ca. 0.06Å). Following the previously devised procedure, we calculated average errors in alanine di- and tetra-peptide conformational energies and backbone angles and found the agreement to be adequate (for example, the alanine tetrapeptide extended-globular conformational energy gap was calculated to be 3.09 kcal/mol quantim mechanically and 3.14 kcal/mol with the POSSIM force field). However, we have now also included simulation of a simple alpha-helix in both gas-phase and water as the ultimate test of the backbone conformational behavior. The resulting alanine and protein backbone force field is currently being employed in further development of the POSSIM fast polarizable force field for proteins.
Figures















References
-
-
(1) See, for example, Caldwell JW, Kollman PA. J Am Chem Soc. 1995;117:4177–4178.Cieplak P, Caldwell J, Kollman P. J Comp Chem. 2001;22:1048–1057.Kaminski GA. J Phys Chem B. 2005;119:5884–5890.Jiao D, Zhang JJ, Duke RE, Li GH, Schneiders MJ, Ren PY. J Comp Chem. 2009;30:1701–1711.Hernandez G, Anderson JS, LeMaster DM. Biochemistry. 2009;48:6482–6494.Wang XY, Zhang JZH. Chem Phys Lett. 2011;501:508–512.
-
-
- Veluraja K, Margulis CJ. J Biomol Struct & Dynamics. 2005;23:101–111. - PubMed
-
-
For representative publications see: Rick SW, Stuart SJ, Berne BJ. J Chem Phys. 1994;101:6141–6156.Liu YP, Kim K, Berne BJ, Friesner RA, Rick SW. J Chem Phys. 1998;108:4739–4755.Ramon JMH, Rios MA. Chem Phys. 1999;250:155–169.Gonzalez MA, Enciso E, Bermejo FJ, Bee M. J Chem Phys. 1999;110:8045–8059.Soetens JC, Jansen G, Millot C. Mol Phys. 1999;96:1003–1012.Dang LX. J Chem Phys. 2000;113:266–273.Chen B, Xing JH, Siepmann JI. J Phys Chem B. 2000;104:2391–2401.Jedlovszky P, Vallauri R. J Chem Phys. 2001;115:3750–3762.Ribeiro MCC. Phys Rev B. 2001;6309:4205.Rinker S, Gunsteren WF. J Chem Phys. 2011;134:084110.Jiang W, Hardy DJ, Phillips JC, MacKerrel AD, Schulten K, Roux B. J Phys Chem Lett. 2011;2:87–92.
-
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous